










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.108 no.4 Madrid abr. 2016
Unusual presentation of Gilbert disease with high levels of unconjugated bilirubin. Report of two cases
Eduardo Flores-Villalba1,2,3, Carlos Rodriguez-Montalvo1,2, Gabriela Arredondo-Saldaña2, Francisco Bosques-Padilla1,2, Tania Zertuche-Maldonado2 and Landy Torre-Flores2
1 Liver Disease Unit. Hospital San Jose-Tec de Monterrey. Monterrey, México.
2 School of Medicine and Health Sciences of Tec Salud. Monterrey, México.
3 Design and Technology Innovation Center of Tecnológico. Monterrey, México
ABSTRACT
Gilbert's syndrome is a benign condition characterized by asymptomatic sporadic episodes of jaundice, due to a mild unconjugated hyperbilirubinemia caused by a deficiency in bilirubin glucoronidation. Under certain physiologic or pathologic events, bilirubin level rises but according to literature it does not reach out more than 3 mg/dl. We report 2 cases of Gilbert's syndrome, genetically tested, which presented with bilirubin levels above 6 mg/dl without any trigger or coexisting condition. In conclusion, bilirubin levels higher than 6 mg/dl in Gilbert syndrome are rare, hemolytic and other metabolism diseases must be ruled out, and genetic testing may be necessary in some cases.
Key words: Gilbert's syndrome. Jaundice. Unconjugated hyperbilirubinemia.
Background
Gilbert syndrome is a benign condition characterized by sporadic episodes of asymptomatic jaundice due to a mild indirect hyperbilirubinemia caused by a deficiency in bilirubin glucoronidation. This condition has been documented in 4 to 16% of general population (1-6). Also named, constitutional hepatic dysfunction, familial nonhemolytic jaundice and Meulengracht disease, Gilbert syndrome, is one of the most common causes of increased unconjugated bilirubin levels besides liver disease, hemolysis and neonatal jaundice.
The normal total bilirubin ranges in a healthy patient varies between 0.0 to 1.0 mg/dL; from which 0.6 to 1 mg/dL correspond to unconjugated bilirubin. Patients with Gilbert syndrome can present fluctuating serum bilirubin levels ranging from normal to usually less than 3 mg/dL. In certain pathologic or physiologic conditions, such as stress, fasting, or coexisting disorders hyperbilirubinemia may rise, however it usually remains below 6 mg/Dl (7-9).
Gilbert's syndrome is caused by a mutation in the proximal promoter of the UGT1A1 gene (10). The natural course of the disease is benign, yet sometimes; diagnosis may not be evident and becomes a challenge when presentation is not typical.
The objective of this article is to report 2 patients that presented with persistent high unconjugated bilirubin levels and where finally diagnosed with Gilbert's syndrome confirmed by genetic analyses.
Case Report
Case report 1
A 17-year-old male presented with a 2-month history of jaundice. Patient was asymptomatic and jaundice was not related to any special activity. On physical examination he had stable vital signs, mild scleral icterus and white skin, abdomen was flat and soft without tenderness and no hepatosplenomegaly was palpable. Initial liver function tests revealed total bilirubin of 6.7 mg/dL with 5.9 mg/dL corresponding to unconjugated bilirubin. Bilirubin levels above 6 mg/dL were confirmed. Remaining results of liver exams were normal as well as blood cell count and chemistry. Exams were solicited in order to exclude other differential diagnosis (Table I).
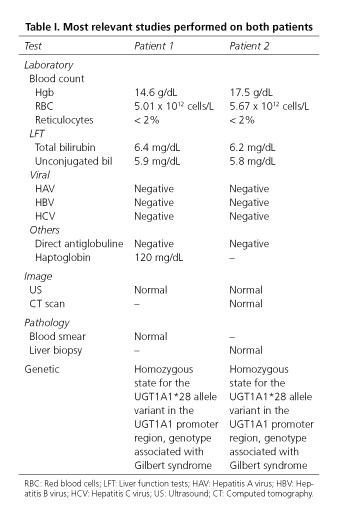
Acute and chronic hepatitis virus panel did not show abnormalities. Autoimmune antibodies were not elevated. No cause of hemolysis was demonstrated. Genetic testing was requested and showed a mutation in UGT1A1 gene, characteristic of Gilbert's syndrome.
Case report 2
A 20-year-old male, first noted with transient hyperbilirubinemia at age of 10. He presented as an outpatient referring jaundice, choluria and fatigue. On physical examination he had stable vitals signs, icterus sclera and generalized jaundice. Rest of the examination was normal. The results of his laboratory exams were 6.2 mg/dL serum total bilirubin with 5.8 mg/dL of unconjugated bilirubin. Other liver function tests were within normal ranges. Workup for unconjugated hyperbilirrubinemia was completed (Table I). Acute and chronic hepatitis virus tests were negative. Antibodies for autoimmune disease were normal. An ultrasonography and CT of liver, gallbladder, pancreas, spleen and both kidneys were normal. A liver biopsy was taken and showed a normal study. The genetic test was positive for Gilbert's syndrome.
Discussion
Gilbert syndrome is characterized by elevated serum bilirubin, specifically unconjugated bilirubin due to a deficiency of bilirubin glucoronidation (11). Indirect hyperbilirubinemia is produced by a defect in the promoter region of the gene that encodes the enzyme responsible of this action, the uridine diphosphoglucoronate-glucoronosyltransferase 1A1(UGT1A1) (4,10). The activity of this enzyme is reduced up to 70% of the normal (11,12).
In both patient's clinical history, a trigger of icterus could not be identified, even during directed questioning. Patients were not under stress conditions when jaundice appeared neither did correlate to any particular activity. Contrary to the typical presentation of Gilbert syndrome, bilirubin levels were persistently above 6 mg/dL, exhibiting an unusual presentation of the disease. As stated before, serum bilirubin rarely exceeds 3 mg/dL even under conditions that exacerbates hyperbilirubinemia in these patients.
In these two cases, although high hyperbilirubinemia was persistent, no other alteration or gene mutation was confirmed, neither in the UGT1A1 gene nor a heterozygous Crigler-Najjar-type structural mutation, an additional coexisting condition that predisposes to hyperbilirubinemia or cause of hemolysis could not be identified (14).
The diagnosis must be suspected when persistent jaundice and elevation of unconjugated hyperbilirubinemia occur in the absence of other causes of indirect hyperbilirubinemia. Other than these findings, normal physical examination, lab exams and imaging studies are the rule, and when biopsy is taken, a normal histopathological liver parenchyma is usually seen (16).
Ordinary genetic test describes a dinucleotide insertion within the TATA box of the promoter region of the UGT1A1 gene. The standard sequence of the gene is A[TA]6TAA, however, in Gilbert syndrome a longer version exists, represented as A[TA]7TAA. This defect is also known as UGT1A1*28 (1,5,10). Genetic tests in both patients confirmed this variant.
Gilbert syndrome does not cause progressive liver damage, or histological changes, it does not lead to hepatic morbidity, and no further treatment or follow up is recommended (17). Under the conditions that presented the disease in these two cases, it was very important to establish and confirm the diagnosis, since other causes of high indirect hyperbilirubinemia require prompt attention and the prognosis may be quite different.
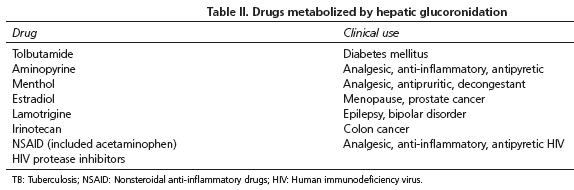
Once diagnosis is made, the most important aspect is to notify the patients on the benign course of the disease and instruct them on the circumstances that may precipitate an elevation of bilirubin and appearance of jaundice. Also is very important to inform the patient on side effects or unexpected toxicity due to some drugs which metabolism is due to hepatic glucoronidation (18) (Table II).
Gilbert's syndrome is an asymptomatic, benign, autosomal recessive disorder characterized by unconjugated hyperbilirubinemia that rarely exceeds 3 mg/dL. However, we present two cases in which it is demonstrated that the diagnosis should not be ruled out even if hyperbilirubinemia exceeds 6 mg/dL. A complete workup, including genetic testing may be necessary in some unusual cases.
References
1. Bosma P, Chowdhury JR, Jansen PH. Genetic inheritance of Gilbert's syndrome. Lancet 1995;346:314-5. DOI: 10.1016/S0140-6736(95)92203-2. [ Links ]
2. Borlak J, Thum T, Landt O, et al. Molecular diagnosis of a familial nonhemolytic hyperbilirubinemia (Gilbert's syndrome) in healthy subjects. Hepatology 2000;32:792-5. DOI: 10.1053/jhep.2000.18193. [ Links ]
3. Monaghan G, Ryan M, Seddon R, et al. Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet 1996;347:578-81. DOI: 10.1016/S0140-6736(96)91273-8. [ Links ]
4. Biondi ML, Turri O, Dilillo D, et al. Contribution of the TATA-box genotype (Gilbert syndrome) to serum bilirubin concentrations in the Italian population. Clinical chemistry 1999;45:897-8. [ Links ]
5. Lampe JW, Bigler J, Horner NK, et al. UDP-glucuronosyltransferase (UGT1A1*28 and UGT1A6*2) polymorphisms in Caucasians and Asians: relationships to serum bilirubin concentrations. Pharmacogenetics 1999;9:341-9. DOI: 10.1097/00008571-199906000-00009. [ Links ]
6. Raijmakers MT, Jansen PL, Steegers EA, et al. Association of human liver bilirubin UDP-glucuronyltransferase activity with a polymorphism in the promoter region of the UGT1A1 gene. J Hepatol 2000;33:348-51. DOI: 10.1016/S0168-8278(00)80268-8. [ Links ]
7. Felsher BF, Rickard D, Redeker AG. The reciprocal relation between caloric intake and the degree of hyperbilirubinemia in Gilbert's syndrome. N Engl J Med 1970;283:170-2. DOI: 10.1056/NEJM197007232830403. [ Links ]
8. Barrett PV. The effect of diet and fasting on the serum bilirubin concentration in the rat. Gastroenterology 1971;60:572-6. [ Links ]
9. Fretzayas A, Moustaki M, Liapi O, et al. Gilbert syndrome. European journal of pediatrics 2012;171:11-5. DOI: 10.1007/s00431-011-1641-0. [ Links ]
10. Koiwai O, Nishizawa M, Hasada K, et al. Gilbert's syndrome is caused by a heterozygous missense mutation in the gene for bilirubin UDP-glucuronosyltransferase. Human molecular genetics 1995;4:1183-6. DOI: 10.1093/hmg/4.7.1183. [ Links ]
11. Black M, Billing BH. Hepatic bilirubin udp-glucuronyl transferase activity in liver disease and gilbert's syndrome. N Engl J Med 1969;280:1266-71. DOI: 10.1056/NEJM196906052802303. [ Links ]
12. Auclair C, Feldmann G, Hakim J, et al. Bilirubin and paranitrophenol glucuronyl transferase activities and ultrastructural aspect of the liver in patients with chronic hemolytic anemias. Biomedicine / (publiee pour l'AAICIG) 1976;25:61-5. [ Links ]
13. Powell LW, Hemingway E, Billing BH, et al. Idiopathic unconjugated hyperbilirubinemia (Gilbert's syndrome). A study of 42 families. N Engl J Med 1967;277:1108-12. DOI: 10.1056/NEJM196711232772102. [ Links ]
14. Ellis E, Wagner M, Lammert F, et al. Successful treatment of severe unconjugated hyperbilirubinemia via induction of UGT1A1 by rifampicin. J Hepatol 2006;44:243-5. DOI: 10.1016/j.jhep.2005.09.011. [ Links ]
15. Muraca M, Fevery J. Influence of sex and sex steroids on bilirubin uridine diphosphate-glucuronosyltransferase activity of rat liver. Gastroenterology 1984;87:308-13. [ Links ]
16. Erlinger S, Arias IM, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology 2014;146:1625-38. DOI: 10.1053/j.gastro.2014.03.047. [ Links ]
17. Lankisch TO, Behrens G, Ehmer U, et al. Gilbert's syndrome and hyperbilirubinemia in protease inhibitor therapy - an extended haplotype of genetic variants increases risk in indinavir treatment. J Hepatol 2009;50:1010-8. DOI: 10.1016/j.jhep.2008.12.030. [ Links ]
18. Burchell B, Soars M, Monaghan G, et al. Drug-mediated toxicity caused by genetic deficiency of UDP-glucuronosyl transferases. Toxicology letters 2000;112-113:333-40. DOI: 10.1016/S0378-4274(99)00209-X. [ Links ]
![]() Correspondence:
Correspondence:
Eduardo Flores-Villalba.
Liver Disease Unit.
Hospital San Jose-Tec de Monterrey.
Ignacio Morones Prieto 3000.
Col. Los Doctores, ZC 64710.
Monterrey, NL, México
e-mail: eduardofloresvillalba@itesm
Received: 04-02-2015
Accepted: 11-04-2015
