










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Cirugía Oral y Maxilofacial
versión On-line ISSN 2173-9161versión impresa ISSN 1130-0558
Rev Esp Cirug Oral y Maxilofac vol.29 no.6 Madrid nov./dic. 2007
CASO CLÍNICO
Tratamiento de la displasia fibrosa asociada a hemofilia C: a propósito de un caso
Treatment for fibrous dysplasia when associated with hemophilia C: A case report
T. Creo Martínez1, A. Borrego Luque2, C. Salazar Fernández2, R. del Rosario Regalado1, Y. Marín Lapeira3
1 Médico Residente. Servicio Cirugía Oral y Maxilofacial.
2 Médico Adjunto. Servicio Cirugía Oral y Maxilofacial.
3 Médico Residente. Servicio Radiodiagnóstico.
Hospital Universitario Virgen Macarena, Sevilla, España
Dirección para correspondencia
RESUMEN
La displasia fibrosa es una enfermedad ósea benigna que cambia el tejido óseo normal por una proliferación de tejido conectivo. Se piensa que la alteración del gen Gsa es la principal razón de la enfermedad. La hemofilia C es una enfermedad sanguínea, hereditaria rara, que provoca hemorragias en pacientes afectos. Es autonómica recesiva, por lo que hombres y mujeres pueden estar afectos.
Paciente de 13 años que desarrolla una displasia fibrosa en maxilar superior derecho que empieza con dolor durante la masticación de alimentos duros. Presenta abombamiento de vestíbulo y enrojecimiento de paladar derecho.
Presenta un déficit discreto de factor XI (heterocigoto). Por ello, necesita una preparación especial antes de extirpar la lesión debido a su déficit. Se ha descubierto que la razón de la displasia fibrosa es la mutación del gen Gsa (GNAS1) que está en el cromosoma 20q.
La causa de la hemofilia C es el déficit del factor XI debido a una mutación del gen FXI en el cromosoma 4. Quizás estas dos raras enfermedades tengan una relación, porque ambas se presentan en el mismo paciente.
Palabras clave: Displasia fibrosa; Hemofilia C; gen Gsa; Pamidronato; Gen FXI.
ABSTRACT
Fibrous Dysplasia is a benign bone disease that changes normal bone tissue for a proliferation of connective fibrous tissue. It is thought that an alteration of the Gsa gene is the main cause of the disease.
Hemophilia C is a rare inherited blood disease leading to abnormal hemorrhages in affected patients. They have a factor XI deficiency. It is the least frequent of all hemophilias. It is a recessive autosomal disease, affecting both men and women. A 13 year-old patient developed fibrous dysplasia in right upper maxilla. The patient started with pain on chewing hard food. She had vestibular swelling and reddening of the right side of the palate. She had a discrete factor XI deficiency (heterozygotic). She needed special preparation before the lesion could be removed because of her deficiency.
It has been discovered that the mutation of gene Gsa (GNAS1) is the reason for fibrous dysplasia. This gene is in chromosome 20q. The cause of hemophilia C is a factor XI deficiency due to a mutation in the FXI gene in chromosome 4. Perhaps these two rare diseases are related, because both are unusual diseases and both are in the same patient.
Key words: Fibrous dysplasia; Hemophilia C; Gene Gsa, Pamidronate; FXI gene.
Introducción
La displasia fibrosa es una patología ósea benigna consistente en el reemplazo progresivo de tejido óseo normal por una proliferación de tejido conectivo fibroso.1 Aunque la etiología no está totalmente esclarecida, se postula que se debe a una alteración del gen Gsa,1,2 debido a una mutación heterocigótica en dicho gen.2 Esta mutación ha sido descrita en la displasia fibrosa monostótica, poliostótica y en la displasia fibrosa asociada al síndrome de McCune-Albright.2
Lichtenstein,5 fue el primero en describir la enfermedad en 1938. La displasia fibrosa puede ser monostótica y poliostótica. La forma monostótica localizada en la región craneofacial sólo supone el 10% de los casos,4 tiene predilección por el maxilar superior,2,3 y puede afectar a huesos adyacentes como el cigomático, esfenoides y occipital.2
La enfermedad aparece en niños, suele progresar durante la pubertad y adolescencia, y en la mayoría de los casos se para al finalizar la adolescencia,6 Hay un grupo de pacientes en los que la enfermedad continúa progresando después de la pubertad, causando deformidad y/o problemas funcionales.6
Se manifiesta en la región facial, bien como crecimiento asintomático del hueso afecto, produciendo deformidad facial, tumefacción de tejidos blandos,7 proptosis, cefaleas, o bien causando dolor de la zona afecta. Puede provocar afectación nerviosa, por compresión de estas estructuras.
La degeneración sarcomatosa puede ocurrir, pero es muy poco frecuente, se estima que ocurre en un 0,5%.8
Radiológicamente se caracteriza por presentar lesiones radiolúcidas, radiotransparentes o radiopacas, escleróticas, dependiendo de la cantidad de componente fibroso. Lo más común es objetivar en la TC una imagen en vidrio esmerilado, o en cáscara de huevo, que corresponde al hueso sano que rodea a la lesión.9
La hemofilia C es una enfermedad hereditaria rara de la sangre, caracterizada por la tendencia a las hemorragias anormales. Es la menos frecuente de todas las formas de hemofilia. Se caracteriza por la deficiencia del factor XI, bien por disminución del factor o por existencia de un factor estructuralmente anormal, (código CIE- 9-MC). La hemofilia C supone el 2-3% de todas las hemofilias, y sus manifestaciones suelen aparecer en el período neonatal porque el factor no atraviesa la placente, aunque puede haber muchos recién nacidos asintomáticos. Los síntomas fundamentales son el hemartros, hematuria, epixtasis etc.10
Es una enfermedad que se hereda como un rasgo genético recesivo incompleto, no ligado al cromosoma X, se encuentra su alteración en los cromosomas autosómicos, por lo que la enfermedad se manifiesta tanto en varones como en mujeres.
Caso clínico
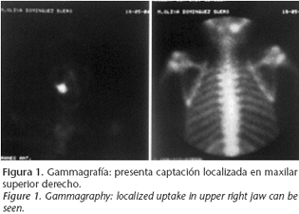
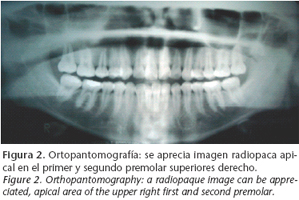
Paciente de 13 años de edad que es remitido a consulta en junio de 2004 al presentar la imagen de captación localizada en el maxilar superior derecho en la gammagrafía (Fig. 1) realizada por dolor lumbar. Niega antecedentes de enfermedades, y a la exploración no se aprecian síntomas intra ni extraorales sugestivos de enfermedad. En la ortopantomografía (Fig. 2), se aprecia imagen radiopaca apical al primer y segundo premolar superiores derechos. En la TC de confirmación diagnóstica se objetiva una imagen en vidrio esmerilado compatible con displasia fibrosa de maxilar superior derecho (Fig. 3). El diagnóstico definitivo se realizó mediante biopsia de la zona afecta.

La paciente, asintomática, se revisa periódicamente, y en septiembre de 2004 comienza con molestias a la masticación de alimentos duros. A la exploración se objetiva abombamiento de vestíbulo y paladar derechos y enrojecimiento de mucosa de revestimiento, sin aumento de la movilidad dentaria. Se propone cirugía con intención de resección completa de la lesión.
En el estudio preoperatorio la analítica detecta una alteración de los parámetros de coagulación. Tras la confirmación se solicita un estudio por el hematólogo, el cual evidencia un déficit discreto del factor XI (heterocigótico) y anemia normocítica normocroma.
La paciente es intervenida en febrero de 2005 bajo anestesia general precisando previamente una preparación prequirúrgica. Se dispone a preparar a la paciente administrándole ácido tranexámico durante la semana previa a la intervención. Doce horas antes de la cirugía se realiza estudio de coagulación encontrándose todos los valores dentro del rango de la normalidad. Se prescribe transfundir dos unidades de plasma fresco treinta minutos antes del acto quirúrgico. Se realizó una hemimaxilectomía derecha (Fig. 4), extirpándose desde tuberosidad del maxilar hasta primer premolar, y hueso palatino, dejando suelo de la órbita. Evolucionó de manera favorable sin presentar complicaciones en el postoperatorio inmediato. Durante el postoperatorio se mantiene el ácido tranexámico y los controles analíticos de coagulación durante siete días. La paciente fue dada de alta a los 10 días sin complicaciones y sin signos de sangrado.

En el postoperatorio temprano presenta dolor en territorio del nervio infraorbitario y anemia no resuelta. En junio del 2005 desaparece la neuralgia, y se hace rehabilitación protésica, mediante prótesis parcial removible, sin presentar problemas ni signos de recidiva.
En agosto del 2005 presenta recidiva a nivel de región malar derecha, estando asintomática. Comienza con dolor localizado en esa zona al mes. Se propone como opción de tratamiento el uso de difosfonatos para paliar el dolor.
Discusión
La displasia fibrosa es una enfermedad benigna, rara, que puede afectar a un solo hueso (tipo monostótico), puede afectar a varios huesos (tipo poliostótico), o puede acompañarse de otra patología sistémica, incluyendo una pigmentación anormal de la piel, prematuridad en el desarrollo sexual e hipertiroidismo (síndrome de McCune-Albrigt). Se puede presentar sola, asociada a otras enfermedades como la enfermedad de Pyle o displasia craniometafisaria.6
En la displasia fibrosa se ha descubierto recientemente como causa de la misma una mutación en el gen Gsa (GNAS1) que se encuentra en el cromosoma 20q11. Esta mutación ocurriría durante el desarrollo embrionario, o durante la vida postnatal. La mutación del gen Gsa produce un aumento de la adenilato ciclasa, lo cual produce un aumento del AMPc intracelular. La alta concentración del AMPc intracelular genera un aumento en la proliferación y diferenciación inapropiada de las células mutadas, causando la formación de una matriz fibrosa inmadura y desorganizada, generando el tejido fibroso de la displasia.2
La hemofila C es una enfermedad rara, debido a la deficiencia del factor XI de la coagulación, que participa en la fase inicial de la vía intrinseca de la coagulación. La mayoría de los déficits de factor XI ocurren en la población judía Ashkenazi.12 Se sabe que la enfermedad es una mutación que ocurre en el gen FXI que se ubica en el cromosoma 4.10 En el gen FXI ocurren tres mutaciones puntuales que dividen la enfermedad en tres tipos según el lugar de mutación que son la tipo I, tipo II y tipo III.13
Nuestro paciente presenta displasia fibrosa monostótica de maxilar superior y hemofilia C heterocigótica, dos enfermedades raras, que en este caso se han presentado conjuntamente en la misma persona, además ambas son dos enfermedades producidas por mutaciones genéticas en genes autosómicos, según los últimos estudios. En función de esto pedimos un cariotipo de la paciente para valorar si presentaba alguna afectación genética conjunta (Fig. 5). El resultado del cariotipo fue que todas las metafases analizadas presentaban 46 cromosomas. Aplicadas técnicas de bandas GTG, no se encontraron anomalías en las metafases estudiadas. Aunque el cariotipo fue normal, no se pudo excluir la posibilidad de algunos mosaicismos cromosómicos, microdeleciones, microduplicaciones, y/o pequeños reordenamientos estructurales.

En la literatura se recoge la relación de la displasia fibrosa con otras enfermedades como la enfermedad de Pyle, sin que todavía esté determinada la relación entre ambos,6 por lo cual nos podemos encontrar ante otra enfermedad que aparece con la displasia fibrosa.
El tratamiento en la displasia fibrosa es muy variable, en función de la sintomatología o la afectación que presenten los pacientes. Así el tratamiento varía desde no hacer nada, controlando al paciente periódicamente, si no tiene sintomatología, hasta hacer resecciones completas del hueso afecto o simplemente realizar curetajes. La elección del tratamiento está en función de la zona afecta y la capacidad de reconstrucción del tejido resecado. Una resección total estaría indicada si el paciente presenta sintomatología como dolor, deformidad facial, o invasión de estructuras importantes, tales como la órbita con compresión de globo ocular o nervio óptico.3,14 Existen en estudio otras posibilidades de tratamiento, que parecen que están ofreciendo resultados alentadores, tal es el caso del uso de pamidronato intravenoso.15,16 El uso de agentes antireabsortivos como los bifosfonatos en el tratamiento de la displasia fibrosa está justificado por los hallazgos histológicos en la displasia fibrosa de un elevado número de osteoclastos, y la evidencia de reabsorción ósea en las lesiones de la displasia fibrosa, así el aumento de la reabsorción ósea parece que juega un papel importante en el establecimento y crecimiento de algunas lesiones de displasia. 16
A nuestra paciente se le ofreció inicialmente un tratamiento quirúrgico de la lesión, además del tratamiento con pamidronato, pero la propia paciente demandó un tratamiento quirúrgico definitivo, (debido al relativo pequeño tamaño de la misma y a que podía ser abordado de manera intraoral). En la preparación prequirúrgica previo a la intervención se detectó hemofilia C, por lo que se tomaron medidas adicionales, aumentando así considerablemente el riesgo de la cirugía.
En la intervención se intentó hacer un tratamiento definitivo, resecando por completo el hueso afecto, con la limitación de ser un abordaje intraoral, pues la paciente se negaba a la realización de un abordaje extraoral, y parecía que mediante una incisión intraoral podía resecarse totalmente la enfermedad, pero meses después de la intervención, la paciente presentó una recaída, con dolor intenso en la zona afecta. Se le planteó otra vez tratamiento quirúrgico, y teniendo en cuenta los elevados riesgos que tiene esta paciente por su enfermedad concomitante y la preparación pre y postquirúrgica que necesita, con los riesgo que implica la transfusión de plasma fresco, y se le planteó por otro lado, tratamiento con pamidronato intravenoso, escogiendo finalmente tratamiento con pamidronato, que en su caso estaría totalmente indicado al ser una displasia fibrosa dolorosa. El pamidronato se administra 1 mg/kg IV durante 3 días cada 4-6 meses. Tras la administración del pamidronato cedieron los dolores en maxilar, estando la paciente libre de sintomatología hasta la fecha.
Uno de los problemas que tiene este tratamiento en niños y adolescentes, es la interferencia con el crecimiento.15 Hay estudios realizados a largo plazo, en los que no hay una gran afectación del crecimiento por los difosfonatos,16 parece que el único efecto secundario registrado durante la perfusión ha sido un aumento de la temperatura corporal en algunos pacientes de 38-40ºC, y que cede sin problema con AINEs.15 Otro efecto secundario registrado ha sido en pacientes con afectación renal (algunos pacientes con síndrome de Albright) evidencia de osteomalacia que cede con la suspensión del tratamiento, y que es evitable en estos pacientes si se asocian los difosfonatos a fósforo por vía oral,16 por lo que se puede considerar el uso de los difosfonatos como un tratamiento seguro.
Por tanto debido a los estudios favorables con este fármaco, su seguridad, y los mayores riesgos que implican en esta paciente un tratamiento quirúrgico más agresivo, se optó por un tratamiento con pamidronato intravenoso.
Aunque el tratamiento quirúrgico de la displasia fibrosa, es una buena opción de tratamiento, parece que en nuestro paciente no es la opción más adecuada, lo que nos demuestra una vez más, que en la displasia fibrosa no se puede hablar de un tratamiento estándar, sino que hay que evaluar los casos individualmente debido a las enfermedades o complicaciones asociadas que puede presentar, y elegir una opción terapéutica u otra en función de cada paciente.
Bibliografía
1. Concha G, Jofré S. Displasia fibrosa en el territorio maxilofacial. Definición e imagen en radiología convencional y TC. Anuario Sociedad de Radiología Oral y Maxilofacial de chile 2003;6-1. [ Links ]
2. Perdigao PF, Pimenta FjGS, Castro WH, De Marco L, Gomez RS. Investigation of the Gsa gen in the diagnostic of fibrous displasia. Int J Oral Maxillofacial surgery 2004;33:498-501. [ Links ]
3. Zenn MR, Zuniga J. Treatment of fibrous dysplasia of the mandible with radical excision and immediate reconstruction: a case report. J Craniofac Surg 2001;12- 3. [ Links ]
4. Gillman G, Bryson PC, Rao UNM. Radiology quitz case 1: Isolated fibrous dysplasia of the sphenoidal sinus. Arch Otolaryngol Head Neck Surgery 2004;130. [ Links ]
5. Lichtenstein L. Polyostotic fibrous dysplasia. Arch Surg 1938;36:874-98. [ Links ]
6. Ozek C, Gundogan H, Bilkay U, Tokat C, Gurler T, Songur E. Craneomaxillofacial fibrous dysplasia. J Craniofac Surg 2002;13-3. [ Links ]
7. Crawford LB. An unusual case of fibrous dysplasia of the maxillary sinus. Am J Orthodontics Dentofac Orthoped 2003;124-6. [ Links ]
8. Michael CP, Lee AG, Patrinely JR, Stal S, Blacklock JB. Visual loss associated with fibrous dysplasia of the anterior skull base. J Neurosurg 2000;92: 350-4. [ Links ]
9. Tokano H, Sugimoto T, Noguchi Y, Kitamura K. Sequential computed tomography images demonstrating characteristic changes in fibrous dysplasia. J Laryngol Otol 2001;115:757-9. [ Links ]
10. Bolton-Maggs PHB. Deficiencia del factor XI y su manejo. Federación mundial de hemofilia. Tratamiento de la Hemofilia 2004;16. [ Links ]
11. Levine MA, Modi WS, O´Brien SJ. Mapping of the gene encoding the alpha subunit of the stimulatory 6 protein of adenylyl cyclase (GNAS1) to 20q13.2-q13.3 in human by in situ hybridization. Genomics 1991;11:478-9. [ Links ]
12. Meijers JCM, Davie EW, Chung DW. Expression of human blood coagulation factor XI: characterization of the defect in factor XI type III deficiency. Blood 1992;79:1435-40. [ Links ]
13. Hancock JF, Wieland K, Pugh RE, Martinowitz U, Schulman S, Kakkar VV, Kernoff PBA, Cooper DN. A molecular genectic study of factor XI deficiency. Blood 1991;77:1942-8. [ Links ]
14. Keijser LCM, Van Tienen TG, Bart Schreuder HW, Lemmens JAM, Pruszczynski M, Veth RPH. Fibrous dysplasia of bone. Management and outcome of 20 cases. J Surg Oncol 2001;76:156-66. [ Links ]
15. Kos M, Luczak K, Godzinski J, Klempous J. Treatment of momostotic fibrous dysplasia with pamidronato. J Craniomaxillofac Surg 2004;32:10-5. [ Links ]
16. Chapurlat RD, Huguensy P, Delmas PD, Meunier PJ. Treatment of fibrous dysplasia of bone with intravenous pamidronate: long term effectiveness and evaluation and evaluation of predictors of response to treatment. Bone 2004; 35:235-42. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Teresa Creo Martínez
Avda. Dr. Fedriani 19, 2º-2º
41009 Sevilla, España
Email: tesicreo@gmail.com
Recibido: 12.04.06
Aceptado: 18.12.06

