










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Cirugía Oral y Maxilofacial
versión On-line ISSN 2173-9161versión impresa ISSN 1130-0558
Rev Esp Cirug Oral y Maxilofac vol.30 no.4 Madrid jul./ago. 2008
Síndrome de hiperparatiroidismo por tumor maxilar
Hyperparathyroidism-jaw tumour syndrome
L. Barroso1, J. Pedro Marcelino2, L. Jiménez Romero1, I. Amado2, A. Ferreira2, C. Alberto Ribeiro3
1 Médico Residente
2 Cirujano Oral y Maxilofacial
3 Jefe de Servicio
Servicio de Cirugía Oral y Maxilofacial. Hospitais da Universidade de Coimbra (HUC), Portugal
Dirección para correspondencia
RESUMEN
El hiperparatiroidismo tiene con frecuencia manifestaciones óseas, de predomínio facial en algunos pacientes.
Los autores describen las manifestaciones en una família de la región central de Portugal, como punto de partida para una revisión de los conocimientos sobre esta entidade clínica todavía poco divulgada y que puede tener como primera manifestación la presentación de tumores faciales.
Palabras clave: Hiperparatiroidismo; Tumores faciales; Fibromas osificantes; Síndrome HPT-TM.
ABSTRACT
Hyperparathyroidism frequently has bone effects. In one subset of patients, these effects involve mainly facial bones (hyperparathyroidism-jaw tumour syndrome).
The authors describe an affected family from central Portugal and discuss the features of this still poorly known disease, which can present initially as a facial tumour.
Key words: Hyperparathyroidism; Jaw tumours; Ossifying fibromas; HPT-JT syndrome.
Introducción
El hiperparatiroidismo primario, con una incidencia que alcanza 1:1000, es la tercera de entre las enfermedades endocrinas más frecuentes, tras la diabetes mellitus y el hipertiroidismo.1 En su mayor parte, los casos son esporádicos y solamente un 10% son familiares.2 El hiperparatiroidismo primario incluye principalmente los síndromes de neoplasia endocrina múltiple de tipo 1 ó 2 (MEN, multiple endocrine neoplasm) y el hiperparatiroidismo asociado a tumor maxilar-el síndrome del hiperparatiroidismo por tumor maxilar (HPT-TM). Se han publicado algunos casos de hiperparatiroidismo familiar aislado pero la mayoría de éstos parecen ser casos menores de los otros síndromes, con un fenotipo incompleto, que probablemente corresponden a mutaciones ligeramente diferentes del mismo gen.3,4
Material y método
En nuestro hospital, se han evaluado y tratado a varias personas que comparten una línea de parentesco común (Fig. 1) y padecen HPT-TM. Esta línea de parentesco ha sido descrita en publicaciones endocrinológicas,5 pero la presentamos aquí a la comunidad de la cirugía oral y maxilofacial para subrayar las manifestaciones faciales implicadas.

Caso 1
En 1993, un varón de 73 años ingresó en el servicio de medicina interna con el diagnóstico provisional de metástasis ósea de tumor primario desconocido. Había perdido 15 kg en 18 meses y manifestaba anorexia, estreñimiento, dolor del muslo izquierdo y elevación de las concentraciones sanguíneas de calcio, bilirrubina y las fosfatasas ácida y alcalina. La gammagrafía ósea (Fig. 2) demostró zonas múltiples de mayor captación y osteoartropatía hipertrófica, sugiriendo enfermedad metastásica.

La historia personal del paciente no ofrecía nada reseñable, salvo haber sido intervenido por un tumor maxilar a la edad de 18 años (1938). No estaba disponible el diagnóstico histológico de este tumor.
Durante el ingreso, se encontraron niveles elevados de parathormona urinaria y de calcio sanguíneo. La ecografía cervical demostró aumento del tamaño de la glándula paratiroidea inferior derecha.
El paciente se sometió a hemitiroidectomía derecha con excisión de la paratiroides agrandada y la glándula ipsilateral; se normalizó inmediatamente la calcemia. El estudio histopatológico demostró carcinoma infiltrante de la paratiroides inferior e hiperplasia de la paratiroides superior.
Dos años más tarde, se presentó de nuevo dolor óseo y estreñimiento con niveles elevados de calcio y de parathormona en sangre. En una nueva intervención, se hizo una hemitiroidectomía, paratiroidectomía izquierda y timectomía. Se normalizó la calcemia pero el paciente murió dos años más tarde por causas no relacionadas.
Caso 2
En 1987, un paciente de 43 años, el hijo mayor del paciente nº 1, ingresó en nuestro servicio de cirugía oral y maxilofacial con un gran tumor de la mandíbula izquierda (Fig. 3).
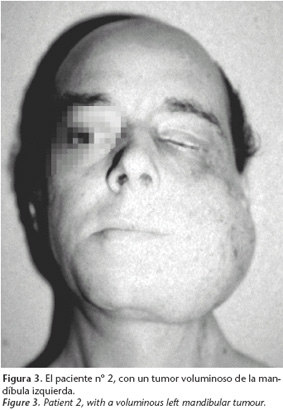
El tumor se había presentado 11 años antes, cuando el paciente vivía en otro lugar del país, y fue diagnosticado en el hospital local como un fibroma osificante. Se detectaron al mismo tiempo niveles altos en sangre de calcio y de parathormona. Se le resecó un adenoma paratiroideo.
Desde entonces, el tumor mandibular seguía creciendo y el paciente fue tratado varias veces con radioterapia y curetaje.
Cuando fue visto por primera vez en nuestro servicio, el paciente tenía un tumor voluminoso de la mandíbula izquierda con zonas de osteorradionecrosis e infección, además de dos masas más pequeñas en la maxila izquierda y la mandíbula derecha (Figs. 4 y 5). Evidenciaba además ceguera izquierda dolorosa producida por la radioterapia e insuficiencia renal incipiente.


Se realizó una enucleación del ojo izquierdo y hemimandibulectomía izquierda; no se hizo la reconstrucción debido a la presencia de infección local. El tumor resecado (13x18 cm) era un fibroma osificante (Fig. 6).

Cinco años más tarde, se realizó hemimandibulectomía derecha con reconstrucción utilizando 2 injertos de costilla (Fig. 7); el diagnóstico histopatológico del tumor derecho fue el mismo. El paciente murió 3 años más tarde por las complicaciones del fallo renal, a pesar de tratarse con hemodiálisis desde 1990.

Caso 3
A la edad de 40 años (1987), este paciente se quejó de dolor de la mano derecha y del tercio medio de la pantorrilla izquierda. En la exploración realizada en el servicio de ortopedia de nuestro hospital, se encontraron lesiones quísticas en estas localizaciones y en una costilla y en la rama mandibular derecha (Fig. 8).

El paciente conocía la existencia de su lesión mandibular desde la edad de 23 años, cuando una biopsia confirmó su benignidad. Desde entonces, apenas había crecido y él no había buscado tratamiento.
Tres años antes, sufrió una fractura patológica de la rótula, que fue tratada en otro hospital.
La lesión cística de la mano derecha se diagnosticó, por biopsia, como osteoclastoma. La presencia de lesiones múltiples se atribuyó a una displasia fibrosa poliostótica.
El año siguiente, el paciente sufrió una nueva fractura patológica del fémur izquierdo (Fig. 9) y varios episodios de hematuria. Durante la evaluación de la hematuria, se encontraron niveles sanguíneos elevados de calcio y de parathormona, además de quistes de la corteza renal.

En 1989, se le hizo una hemitiroidectomía y se resecó un adenoma de la paratiroides inferior derecha.
Desde entonces, han permanecido normales los niveles de calcio y parathormona en sangre y el tumor mandibular no ha aumentado. El paciente no desea realizar una resección de esta lesión.
Caso 4
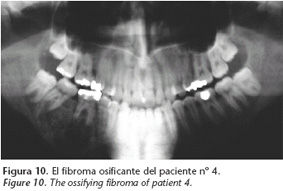
Estudiamos a esta paciente, hija del paciente nº 2, en nuestro servicio en 1995, a la edad de 21 años. Presentó un tumor bien delimitado del cuarto cuadrante (Fig. 10), que extendía hasta el reborde basilar y tenía aspecto quístico, pero con cierta trabeculación.
Debido a sus antecedentes familiares de hiperparatiroidismo y tumor maxilar, se le remitió al servicio de endocrinología para evaluación.
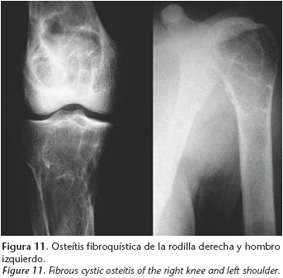
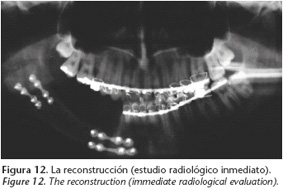
Se encontraron niveles elevados de calcio y parathormona en sangre y osteítis fibroquística en estado avanzado (Fig. 11). El adenoma paratiroideo fue resecado, seguido más adelante por hemimandibulectomía derecha segmentaria con reconstrucción utilizando un aloinjerto de cadáver (Fig. 12). El tumor era un fibroma osificante.
Caso 5
Esta paciente, hija del paciente nº 3, fue el objeto de un estudio de detección bioquímica y radiológica a la edad de 14 años. Se encontró hipocalcemia, hiperparatiroidismo, dos tumores maxilares (de los cuadrantes segundo y cuarto) y varios quistes renales
Se realizó paratiroidectomía derecha para tratar un adenoma paratiroideo. La calcemia permaneció normal durante un año, pero a partir de entonces empezó a aumentar. La paciente se intervino de nuevo (paratiroidectomía izquierda, tanto superior como inferior, y excisión de la mitad de la paratiroides superior derecha por un adenoma de la paratiroides superior izquierda). Las lesiones maxilares seguían creciendo y unos meses más tarde fueron resecados los dos fibromas osificantes (Fig. 13).

Posteriormente tuvo otro episodio de hiperparatiroidismo y fue intervenida para resecar el adenoma de la glándula restante. No hay ningún indicio de recidiva o de la presentación de tumores maxilares nuevos.
Discusión
En 1958, Jackson,6 describió a una familia con hiperparatiroidismo hereditario, en la mayoría de los casos acompañado de tumor maxilar, después de publicarse las observaciones de otros autores, en que definieron este síndrome,7 y lo diferenciaron de la neoplasia endocrina múltiple de tipos 1 y 2 (MEN-1 y MEN-2).8
El HPT-TM es una enfermedad hereditaria autosómica dominante relacionada con el cromosoma 1 (1q25-q31),9 que origina tumores paratiroideos, fibroma osificante de la maxila, diversas lesiones renales (quistes, el tumor de Wilms, hamartoma, adenoma cortical, carcinoma de células papilares) y otros tumores (adenocarcinoma pancreático, tumor testicular, tumor de células de Hürthle del tiroides, pólipos adenomiomatosos del útero).10-12,18
Las manifestaciones más frecuentes (adenomas paratiroideos y fibromas osificantes) se presentan a edades más jóvenes que en los casos esporádicos, que son más frecuentes a la edad media,12 y después de la tercera década de la vida,13 respectivamente.
El hiperparatiroidismo es de origen tumoral, no de hiperplasia paratiroidea, como sucede en los síndromes de neoplasia endocrina múltiple. En el HPTTM, los adenomas a menudo son quísticos; la incidencia de carcinoma es del 5%, mucho más alta que la incidencia de este cáncer en la población general (1:5.000.000).11,14,15 No es infrecuente que un paciente desarrolle varios adenomas tras resecar uno,8 como sucedió en los pacientes 1 y 5.
El fibroma osificante está presente en aproximadamente la mitad de los pacientes y su comportamiento es independiente de la calcemia.8,9 Está bien delimitado e inicialmente radiotransparente, pero se calcifica es progresivamente. Puede desplazar o, raramente, destruir las raíces dentarias. En las imágenes radiológicas puede asemejarse a la displasia fibrosa, pero la displasia fibrosa no tiene bordes definidos y no destruye las raíces dentarias. Su aspecto histológico es de hueso en un estroma fibrocelular, con zonas de hueso laminar y, a veces, cemento. A diferencia, el hueso laminar y el cemento no aparecen en la displasia fibrosa.
En los pacientes que tienen hiperparatiroidismo, el fibroma osificante puede coexistir con la osteítis fibroquística, que origina lesiones sobre todo radiotransparentes y multifocales, y que tiene un aspecto histológico de fibrosis, hemorragia medular y abundantes células gigantes. Estas lesiones, también conocidas como tumores pardos, tienen un aspecto histológico que es difícil distinguir del aspecto de los tumores y granulomas de células gigantes, el quiste aneurismático de hueso e, incluso, la displasia fibrosa (como ocurrió con el paciente nº 3), aunque son mucho menos numerosas las células gigantes en la displasia fibrosa. El hiperparatiroidismo debe descartarse cuando se encuentran estos hallazgos en una biopsia, especialmente si las lesiones son múltiples.
En este grupo de pacientes emparentados, los tumores maxilares parecían tener una mayor penetrancia que el 50% habitual, ya que 5 de los 7 pacientes con la anomalía genética tuvieron fibromas maxilares. Estos tumores se presentaron a la edad típica (de 14 a 32 años) y evidenciaron un comportamiento clínico variable: ninguna recidiva en los pacientes 1, 4 y 5 (durante períodos de 58, 7 y 3 años), crecimiento muy lento durante más de 30 años en el paciente nº 3 y crecimiento persistente multifocal en el paciente nº 2.
El hiperparatiroidismo se manifestó de varias maneras: simulando metástasis óseas (paciente nº 1), originando nefrolitiasis y fracturas patológicas (paciente nº 3), con silencio clínico y lesiones óseas extensas (paciente nº 4) o únicamente como alteración bioquímica (paciente nº 5). Cada uno de estos pacientes, excepto el nº 1, era mucho más joven que el paciente habitual con hiperparatiroidismo esporádico.
Ésta es una alteración rara que puede presentarse inicialmente como tumor maxilar. Los especialistas en cirugía oral y maxilofacial deben conocer su existencia con el fin de descartar el hiperparatiroidismo en pacientes que presentan fibroma osificante, como ya hacen muchos cirujanos en los casos de granuloma o un tumor de células gigantes y de displasia fibrosa multifocal.
En los pacientes afectados, se debe tratar el hiperparatiroidismo y explorar los otros órganos, incluido el área maxilofacial, para detectar fibromas osificantes porque estos pueden recurrir, incluso después de normalizarse la calcemia. Se les debe ofrecer a las familias asesoramiento genético.
Bibliografía
1. Aurbach G, Marx S, Spiegel A. Parathyroid hormone, calcitonin and calciferols. En: Wilson JD, Foster D ed. Textbook of Endocrinology, 7ª ed. Filadelfia, WB Saunders 1992:1397-476. [ Links ]
2. Heath H, Hobbs M. Familial hyperparathyroid syndromes. En: Favus MJ, ed. Primer on Metabolic Bone Disorders and Disorders of Mineral Metabolism. 3ª ed Filadelfia, Lippincott-Raven 1996;187-9. [ Links ]
3. Teh BT, Farnebo F, Twigg S, Höög A y cols. Familial isolated hyperparathyroidism maps to the hyperparathyroidism-jaw tumor locus in 1q21-q32 in a subset of families. J Clin Endocrinol Metab 1998;83:2214-20. [ Links ]
4. Cetani F, Pardi E, Giovannetti A, Vignali E y cols. Genetic analysis of the MEN1 gene and HPRT2 locus in two Italian kindreds with familial isolated hyperparathyroidism. Clin Endocrinol 2002;56:457-64. [ Links ]
5. Cavaco BM, Barros L, Pannett AAJ, Ruas L y cols. The hyperparathyroidism-jaw tumour syndrome in a Portuguese kindred. Q J Med 2001;94:213-22. [ Links ]
6. Jackson CE. Hereditary hyperparathyroidism associated with recurrent pancreatitis. Ann Intern Med 1958;49:829-36. [ Links ]
7. Rosen IB, Palmer JÁ. Fibroosseous tumors of the facial skeleton in association with primary hyperparathyroidism: an endocrine syndrome or coincidence? Am J Surg 1981;142:494-8. [ Links ]
8. Jackson CE, Norum RA, Boyd SB, Talpos GB y cols. Hereditary hyperparathyroidism and multiple ossifying jaw fibromas: a clinically and genetically distinct syndrome. Surgery 1990;108:1006-13. [ Links ]
9. Szabo J, Heath B, Hill V, Jackson C y cols. Hereditary hyperparathyroidism- jaw tumour syndrome: The endocrine tumour gene HRPT2 maps to chromosome 1q21-q31. Am J Hum Genet 1995;56:944-50. [ Links ]
10. Teh BT, Farnebo F, Kristoffersson U, Sundelin B y cols. Autosomal dominant primary hyperparathyroidism and jaw tumour syndrome associated with renal hamartomas and cystic kidney disease: linkage to 1q21-q32 and loss of the wild type allele in renal hamartomas. J Clin Endocrinol Metab 1996;81:4204-11. [ Links ]
11. Dinnen JS, Greenwood RH, Jones JH, Walker DA y cols. Parathyroid carcinoma in familial hyperparathyroidism. J Clin Pathol 1977;30:966-75. [ Links ]
12. Haven CJ, Wong FK, van Dam EW, Van der Juijt y cols. A genotypic and histopathological study of a large Dutch kindred with hyperparathyroidism- jaw tumor syndrome. J Clin Endocrinol Metab 2000;85:1449-54. [ Links ]
13. Eversole LR, Leider AS, Nelson K. Ossifying fibroma: a clinicopathologic study of sixty-four cases. Oral Surg Oral Med Oral Pathol 1985;60:505-11. [ Links ]
14. Streeten EA, Weinstein LS, Norton JA y cols. Studies in a kindred with parathyroid carcinoma. J Clin Endocrinol Metab 1992;75:362-6. [ Links ]
15. Wassif WS, Moniz CF, Friedman E, Wong S y cols. Familial isolated hyperparathyroidism: a distinct genetic entity with an increased risk of parathyroid cancer. J Clin Endocrinol Metab 1993;77:1485-9. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Leonor Barroso
Serviço de Cirurgia Maxilo-Facial
Hospitais da Universidade de Coimbra
3000-075 Coimbra, Portugal
Email: leonorbarroso@netcabo.pt
Recibido: 24.11.05
Aceptado: 16.06.08

