










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Cirugía Oral y Maxilofacial
versão On-line ISSN 2173-9161versão impressa ISSN 1130-0558
Rev Esp Cirug Oral y Maxilofac vol.36 no.2 Madrid Abr./Jun. 2014
https://dx.doi.org/10.1016/j.maxilo.2012.04.006
CASO CLÍNICO
Fibrodisplasia osificante progresiva. El papel del cirujano oral y maxilofacial. Experiencia en 2 casos
Fibrodysplasia ossificans progressiva. The role of oral and maxillofacial surgeon. Experience in two cases
Guillermo García-Serrano, Kora Sagüillo, Fernando Almeida, Jorge Núñez y Manuel Picón
Servicio de Cirugía Oral y Maxilofacial, Hospital Universitario Ramón y Cajal, Madrid, España
Dirección para correspondencia
RESUMEN
La fibrodisplasia osificante progresiva es un raro desorden genético que se caracteriza por la asociación de malformaciones congénitas y brotes de osificación heterotópica progresiva desde los primeros años de vida.
El escaso número de pacientes que padecen la enfermedad, hace que el curso y tratamiento de la misma sea desconocida para muchos especialistas, pese a la importancia del manejo multidisciplinar de estos pacientes.
Se presentan 2 casos valorados por nuestro servicio en los últimos 12 meses; 2 varones de 40 y 23 años diagnosticados de fibrodisplasia osificante progresiva en la infancia que acudieron a la consulta de cirugía maxilofacial para valoración y tratamiento.
Palabras clave: Fibrodisplasia osificante progresiva. Conectivopatías congénitas. Proteína morfogenética ósea.
ABSTRACT
The ossificans progressive fibrodysplasia is a rare genetic disorder that is characterized by the association of congenital malformations and progressive heterotopic ossification outbreaks since the early years of life.
The scanty number of patients who suffer the disease, does that the course and treatment of the same one is not known for many specialists, in spite of the importance of the multidisciplinary managing of these patients. We present 2 cases evaluated by our service in the last 12 months; two 40 and 23-year-old males diagnosed in childhood with ossificans progressive fibrodysplasia who attended the maxilofacial surgery department for evaluation and treatment.
Keywords: Progressive fibrodysplasia ossificans. Congenital connective tissue disorders. Bone morphogenetic protein.
La fibrodisplasia osificante progresiva idiopática (FOPI) es un raro desorden genético que se encuentra dentro del heterogéneo grupo de trastornos del tejido conectivo1.
Esta enfermedad se caracteriza por la asociación de malformaciones congénitas y brotes de osificación heterotópica progresiva desde los primeros años de vida.
La prevalencia de la enfermedad es de un caso por cada 1.640.000 habitantes en el Reino Unido, con menos de 200 casos diagnosticados en el mundo.
Se trata de una enfermedad con herencia AD, aunque la gran mayoría de los casos se derivan de una mutación espontánea en el gen que regula la expresión de BMP (Bone morphogenetic protein) esta proteína actúa como osteoinductora, dirigiendo la diferenciación osteoblástica2.
En 2009 se descifró por completo el mapa genético de la enfermedad, localizando la mutación en el gen ACVR1 en el brazo largo del cromosoma 4 y encargado de la síntesis de BMP3. El efecto de la mutación consiste en una sobreproducción de la proteína al activarse la respuesta inflamatoria del organismo. De forma que se produce una respuesta osteoinductora en regiones donde tan solo debería producirse una respuesta inflamatoria. Las consecuencias son brotes de osificación heterotópica que aparecen desde los primeros años de vida ante cualquier proceso inflamatorio, desde inyecciones intramusculares hasta grandes traumatismos.
De manera característica la enfermedad respeta la musculatura lisa y cardiaca, además de músculos oculomotores, diafragma y lengua; y tiene especial predilección por la musculatura axial, siendo la paraespinal la primera que se afecta en la mayoría de los casos. Los pacientes quedan cada vez más incapacitados, con más limitaciones funcionales y mayores secuelas estéticas a medida que se suceden los brotes.
El diagnóstico de sospecha se obtiene desde el nacimiento por la malformación característica que acompaña a estos niños y que consiste en un primer dedo del pie hipoplásico y que asocia hallux valgus4. Y se confirma con los episodios de osificación de partes blandas que se inician en la infancia5. Los parámetros analíticos no muestran alteraciones, a excepción de la fosfatasa alcalina que aparece elevada en exceso.
A medida que se suceden los brotes, pueden observarse tractos osteofibrosos radiopacos en los músculos afectos.
Aunque la biopsia de estas lesiones está completamente contraindicada por tratarse de un procedimiento traumático que puede desencadenar un brote, los estudios histológicos muestran al inicio de las lesiones áreas extensas de fibroblastos junto a zonas de sobre zonas de destrucción de fibras musculares e infiltración del tejido celular subcutáneo por gran número de células mononucleares. En el centro de este conglomerado de tejido fibroso podemos encontrar áreas con tejido osteocartilaginoso. Cuando las lesiones están ya desarrolladas podemos observar hueso maduro incluso con sistemas haversianos completamente desarrollados6.
El plan de vida de estos pacientes es muy desalentador ya que desarrollan una incapacidad progresiva y permanente mientras son personas jóvenes. La esperanza de vida media se sitúa alrededor de los 40 años, siendo muy pocos los que superan la cifra de 50 años. La principal causa de fallecimiento es un síndrome de insuficiencia torácica que lleva la parada cardiorrespiratoria7.
El tratamiento permanece en periodo de investigación sin que exista actualmente un tratamiento curativo de la enfermedad. Se ha observado que los bifosfonatos consiguen reducir ligeramente el número de brotes y que el uso de antiinflamatorios corticoideos consigue atenuar las consecuencias de los mismos8. Sin embargo, la prevención de cualquier traumatismo o proceso inflamatorio continúa siendo la parte más importante del tratamiento. Las últimas investigaciones parecen dirigir los objetivos terapéuticos a encontrar una diana genética que permita evitar la sobreproducción de BMP.
Pacientes y método
Durante el último año hemos valorado en nuestro servicio a 2 pacientes con FOPI. A raíz de la llegada de estos pacientes se realizó una recogida de datos y recopilación de artículos relacionados con la FOPI, con el objetivo de conocer algo más de esta patología. Para ello se usó la base de datos Pubmed introduciendo los términos:
Fibrodysplasia ossificans progressiva, connective tissue disorders, bone morphogenetic protein
El primero de ellos se trataba de un varón de 41 años que acudió a las consultas de cirugía oral remitido desde el servicio de Reumatología para valoración de las opciones terapéuticas de un trismus en progresión desde hace varios años.
Se trataba de un paciente con una larga evolución de la enfermedad e ingresos repetidos desde la infancia para tratamiento de brotes secundarios a procesos inflamatorios derivados en la mayoría de los casos de traumatismos. No presentaba alergias conocidas, ni otros antecedentes de interés.
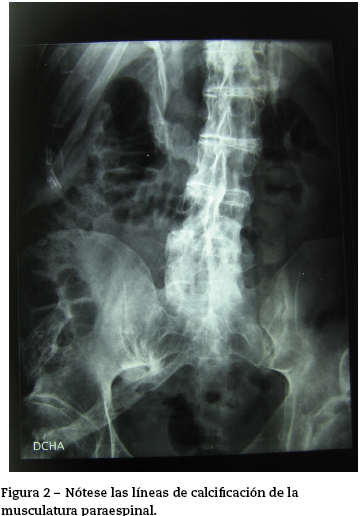
En el momento de la consulta no presentaba síntomas que evidenciaran un brote agudo, sin embargo, eran patentes las secuelas producidas por la enfermedad. El paciente se encontraba en silla de ruedas por la gran limitación en la estática y en la funcionalidad producida por calcificación a nivel de la musculatura y resto de partes blandas, de predominio axial (figs. 1 y 2). En tratamiento con ciclos semestrales de pamidronato y rescate de los brotes con corticoterapia.

A nivel del área maxilofacial, presentaba hipomimia y gran limitación de la apertura oral, con una distancia interincisal de apenas 1 mm (fig. 3). A la palpación se encontraba un tejido indurado en la región de ambas ATM. En la OPG se observaron tractos de osificación entre las superficies articulares a nivel bilateral.

Se comentó con el paciente la imposibilidad de tratamiento quirúrgico, dadas las características de su patología, ya que cualquier actitud intervencionista produciría un agravamiento de las secuelas por aumento de las zonas traumatizadas y por tanto con riesgo de osificación. Fue dado de alta con recomendaciones.
El segundo paciente se trataba de un paciente de 21 años diagnosticado de la enfermedad a los 4 años de edad cuando sufrió el primer brote. En este caso se trató de una interconsulta desde el servicio de Reumatología donde el paciente permanecía ingresado por un cuadro de inflamación a nivel submandibular y submental.
No presentaba alergias conocidas y displasia en la cadera derecha como único antecedente de interés.
Dada la edad del paciente y el diagnóstico temprano de su patología, no presentaba grandes secuelas, ni limitaciones. La afectación se limitaba a una desviación de la columna cervical por fusión parcial vertebral y limitación en la movilidad del hombro izquierdo donde sufrió un brote postraumático en los meses previos.
En tratamiento con ciclos trimestrales de pamidronato y corticosteroides en brotes agudos. Habiendo completado 7 ciclos con escasa mejoría de la clínica.
En el área maxilofacial presentaba una buena apertura oral de alrededor de 35 mm con ligera hipomotilidad a nivel de ATM izquierda en relación con tracto de osificación a ese nivel. (fig. 4).

En la región submental y parte de la región submandibular hasta hioides se palpaban todos los tejidos blandos indurados y aumentados de tamaño, sin que fuera posible la identificación de estructuras a la palpación. Se diagnosticó como un brote agudo de la enfermedad y se trató con corticoides a dosis altas con franca mejoría del cuadro agudo. Fue dado de alta con una pauta corticoidea descendente. En el seguimiento a los 3 meses no se objetiva secuela alguna del brote a nivel submandibular ni submental.
Dadas las características de la enfermedad, se descartó en ambos casos cualquier procedimiento intervencionista y se indicó tratamiento médico y medidas de prevención de traumatismos y de mantenimiento de correcta salud buco dental, con el objetivo de evitar patología que pudiera requerir cualquier tipo de intervención por parte del cirujano maxilofacial.
Discusión
La FOPI es una enfermedad que rara vez veremos a lo largo de nuestra práctica clínica debido a su baja frecuencia. Sin embargo, conviene tener unas nociones básicas de la enfermedad y sobre todo conocer los riesgos de un tratamiento quirúrgico en estos pacientes.
En la mayoría de los casos el cirujano maxilofacial servirá de consultor para especialidades que se ocupan del tratamiento de base y el seguimiento de estos pacientes, sobre todo reumatólogos y rehabilitadores8.
Dicho tratamiento se basa en los escasos hallazgos encontrados en los pocos estudios que se han realizado de una enfermedad en la que la baja prevalencia no invita a grandes inversiones en su investigación. Se ha demostrado la efectividad de las pautas de corticoides intravenosos a altas dosis para los brotes de la enfermedad. Otros antiinflamatorios de menor potencia también serán efectivos aunque con resultados más pobres. Sin embargo, aún no se ha encontrado un tratamiento de base de la enfermedad, una terapia que consiga evitar la aparición de brotes y por tanto sus invalidantes secuelas. Además, en los últimos años el uso de ciclos de bifosfonatos ha demostrado reducir el número de brotes de la enfermedad pero los resultados a largo plazo de los diversos estudios siguen siendo muy limitados.
El gran desarrollo del la ingeniería genética de los últimos años, ha permitido recientemente el mapeo de la enfermedad y parece encaminar la búsqueda hacia una diana terapéutica genética sobre la que podamos actuar para evitar la expresión de la proteína causante9. Sin embargo, dada la escasez de estudios en marcha no se esperan resultados concluyentes a medio plazo.
Al parecer el tratamiento más efectivo en la actualidad estaría basado en las medidas preventivas destinadas a minimizar las situaciones en las que los tejidos están sometidos a procesos inflamatorios. Para ello, el consejo multidisciplinar y un correcto sistema de asistencia de estos pacientes cuando ya sufren secuelas funcionales serán tremendamente útiles.
En caso del cirujano maxilofacial, convendrá minimizar los traumatismos localizados en el área maxilofacial, así como orientar al paciente en los cuidados de salud bucodental y cuidados articulares de las articulaciones temporomandibulares. Indicar revisiones odontológicas periódicas y una correcta higiene oral10.
Todo debe quedar englobado en un enfoque multidisciplinar del tratamiento, en el que trabajen de forma coordinada y conjunta todos los especialistas que pueden aportar una mejoría en la calidad de vida y endentecer la progresión de la enfermedad: traumatólogos, rehabilitadores, reumatólogos, fisioterapeutas, neumólogos, etc.
Conclusiones
Solemos estar acostumbrados a que se recurra a la figura del cirujano cuando hay que dar un paso más en el tratamiento de las patologías y adquirir una actitud intervencionista. Sin embargo, el papel de los cirujanos en el manejo de la FOPI, ha de ser distinto, puesto que deben aplicar sus conocimientos estructurales y fisiopatológicos con el objetivo de prevenir el inicio de los problemas y evitar así la necesidad de futuras intervenciones más agresivas.
Este es sin duda un papel al que los cirujanos no estamos acostumbrados y aún menos en situaciones en las que vemos a pacientes jóvenes que se ven tremendamente invalidados por cuadros que resolvemos diariamente de forma quirúrgica y que sin embargo tienen absolutamente contraindicada la cirugía. Esta es la paradoja de esta enfermedad y el motivo de presentarla en un contexto quirúrgico.
Responsabilidades éticas
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. Kaplan FS, Shore EM, Connor JM. Fibrodysplasia ossificans progressiva (FOP). Wiley-Liss; John Wiley & Sons; Inc, (2002) pp. 827-840. [ Links ]
2. Kaplan FS, McCluskey W, Hahn G, Tabas J, Muenke M, Zasloff MA. Genetic transmission of fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 1993;75:1214-20. [ Links ]
3. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genet. 2006;38:525-7. [ Links ]
4. Kaplan FS, Glaser DL, Shore EM, Deirmengian P, Gupta R, Delai P, et al. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:183-8. [ Links ]
5. Mahboubi S, Glaser DL, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva. Pediatr Radiol. 2001;31:307-14. [ Links ]
6. Kaplan FS, Tabas J, Gannon FH, Finkel G, Hahn GV, Zasloff MA. The histopathology of fibrodysplasia ossificans progressiva: an endochondral process. J Bone Joint Surg Am. 1993;75:220-30. [ Links ]
7. Kaplan FS, Glaser DL. Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:213-6. [ Links ]
8. Glaser DL, Kaplan FS. Treatment considerations for the management of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:243-50. [ Links ]
9. Kaplan FS, Glaser DL, Pignolo RJ, Shore EM. A new era for fibrodysplasia ossificans progressiva: a druggable target for the second skeleton. Expert Opin Biol Ther. 2007;7:705-12. [ Links ]
10. Nussbaum BL, Grunwald Z, Kaplan FS. Oral and dental healthcare and anesthesia for persons with fibrodysplasia ossificans progressiva. Ilin Rev Bone Miner Metab. 2005;3:239-42. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
maxiloramonycajal@gmail.com (G. García-Serrano)
Recibido el 28 de marzo de 2012
Aceptado el 24 de abril de 2012

