










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNeurocirugía
versión impresa ISSN 1130-1473
Neurocirugía vol.20 no.1 feb. 2009
Childhood spinal tumours
F.J. Villarejo; J.M. Belinchón; R. Alfaro; C. Pérez; B. Rivero; A. Pascual y F. Cordobés
Servicio de Neurocirugía. Hospital Infantil Universitario Niño Jesús. Madrid.
Dirección para correspondencia
RESUMEN
Presentamos una extensa serie de 48 tumores espinales en niños diagnosticados y tratados en nuestro centro en el período 1986-2006. Los tumores espinales infantiles suponen una patología infrecuente y heterogénea que es habitualmente diagnosticada tarde por la inespecificidad del cuadro clínico. Los síntomas suelen limitarse a dolor difuso de espalda o deformidades espinales meses antes de debutar con parálisis de miembros inferiores o trastornos esfinterianos. El diagnóstico se realiza con TAC o RM. En la mayoría de casos el tratamiento es quirúrgico. El pronóstico de estos tumores es variable dependiendo de la histología de cada lesión, pudiendo requerir tratamiento complementario con radioterapia y/o quimioterapia.
Palabras clave: Tumor extradural. Fijación espinal. Tumor intradural extramedular. Tumor intramedular. Tumor espinal.
SUMMARY
We report a large series of 48 childhood spine tumors diagnosed and treated at our Hospital between 1986 and 2006. Spinal tumors in children are a rare and heterogeneous condition that frequently are diagnosed late because of their uncharacteristic clinical picture. Symptoms are usually limited to difuse backpain or spinal deformities, prior to leg paresis or sphinter dysfunction. Diagnosis is usually made with MRI or CT. Treatment is surgical in most cases. The prognosis is variable due to the diverse histological findings and it may require the use of complementary treatments as chemoterapy or radiation.
Key words: Extradural tumor. Intradural extramedullar tumor. Intramedullar tumor. Spinal fixation. Spinal tumor.
Introducción
Los tumores espinales infantiles suponen una patología poco frecuente, con una incidencia anual de 1 caso por millón de niños1. La mayoría de los casos se presentan de modo insidioso con dolor local o referido y manifestaciones musculoesqueléticas inespecíficas, por lo que en muchos pacientes se retrasa el diagnóstico con distintas pruebas, tratamientos conservadores o diagnósticos erróneos2-5.
Resulta difícil establecer protocolos de actuación debido a la gran diversidad histológica posible, por lo que los pilares básicos del tratamiento serían la exéresis completa cuando es posible, tratamiento quimio-radioterápico adyuvante según histología e intento de preservación de la función neurológica. En algunos casos debe considerarse la estabilidad de la columna en crecimiento6,7.
El pronóstico, tanto vital como funcional, depende de la histología, agresividad quirúrgica y diagnóstico precoz5,8. La prueba diagnóstica de elección es la resonancia magnética, que además podrá orientarnos sobre la probable histología según localización y secuencia de imágenes9.
Material y métodos
Se han revisado las historias de 48 niños con tumores raquimedulares tratados en nuestro centro en el período 1986-2006. Se incluyeron lesiones pseudotumorales potencialmente compresivas como dos hemangiomas cavernosos que sangraron, un quiste aracnoideo y un quiste hidromiélico. Se excluyeron lesiones asociadas a disrafismos como lipomas o quistes epidermoides de cono medular.
Se analizaron en este estudio edad, sexo, clínica de debut, retraso diagnóstico, localización y extensión del tumor, pruebas diagnósticas, manejo terapéutico y evolución.
Resultados
Nuestra casuística comprende 48 casos, con una edad media de 6,1 años (rango 1 mes-16 años), de los que 18 fueron niñas (37,5%) y 30 niños (62,5%), con una relación próxima a 2:1 a favor de los varones.
1. Diagnóstico histológico
Los tumores más frecuentes fueron de naturaleza benigna, representando 2/3 del total (32 casos). El tercio restante correspondió a tumores malignos (16 casos). La distribución de edad fue similar en ambos grupos, 6,3 años para los benignos y 6,5 para los malignos. En este último subgrupo, si se prescinde del neuroblastoma (dada su temprana aparición en lactantes) la edad media ascendió a 11,4 años.
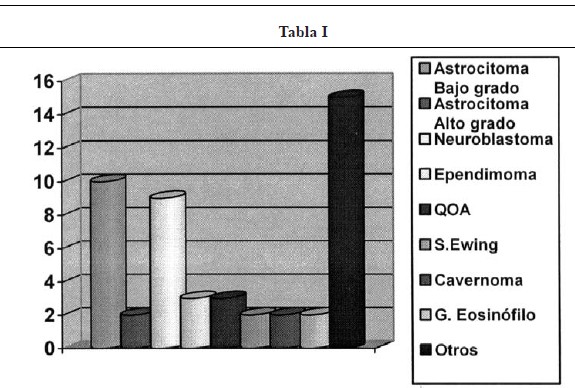
El grupo mayoritario lo constituyeron los astrocitomas (Figs. 2, 3 y 4), con un total de 12 pacientes (25%), de los que 10 fueron de bajo grado y 2 de alto grado (astrocitoma anaplásico y glioblastoma multiforme). En segundo lugar, los neuroblastomas, que supusieron 9 casos (18,7%), de presentación casi exclusiva en lactantes. El tercer grupo correspondió a 3 casos de ependimoma y 3 quistes óseos aneurismáticos (6,25% cada grupo). Además hubo dos casos de sarcoma de Ewing, cavernoma, Schwannoma y granuloma eosinófilo. Finalmente casos aislados de ganglioglioma, hemangioblastoma (Fig 1), lipoma, meningioma, neurofibroma, osteoma osteoide, osteosarcoma, cordoma, quiste siringomiélico, quiste dermoide, quiste aracnoideo y compresión medular por vértebra D6 hemangiomatosa. (Tabla I).

Figura 1. IRM T1 sagital: hemengioblastoma dorsal.
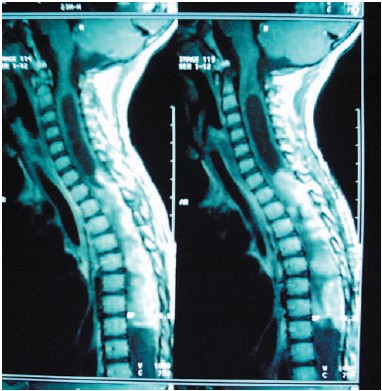
Figura 2. IRM T1 sagital: astrocitoma cervicodorsal.
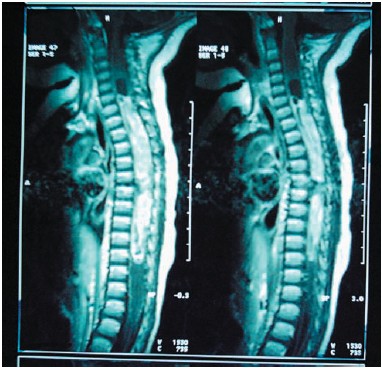
Figura 3. IRM T1 sagital: astrocitoma cervicodorsal

Figura 4. IRM T1 axial: astrocitoma dorsal.
2. Localización de las lesiones
La localización más frecuente de las lesiones fue la columna dorsal, con 23 casos (48%) seguida de la columna cervical, con 9 casos (18,7%). El resto se distribuyeron por todo el eje espinal, desde la unión bulbo-medular a la columna lumbar. Los astrocitomas presentaron la mayor extensión, con una media de 8 segmentos afectos, e incluso un caso de localización de bulbo a cono medular. (Tabla II).

En 17 casos la lesión fue de localización intramedular (35,4%), casi todos fueron astrocitomas; 9 casos fueron intradurales extramedulares (18,7%) y 22 casos fueron extradurales (45,8%). En este último subgrupo la mayoría corresponde a lesiones óseas o de vecindad con invasión de canal, siendo exclusivamente epidurales (sin invasión desde hueso) los cavernomas y el schwannoma. (Tabla III).

3. Presentación y cuadro clínico
Los síntomas y evolución previa al diagnóstico fueron muy variables, con una duración media de 9,4 meses (rango 1 día-11 años). 8 pacientes (16,6%) debutaron con clínica aguda, consistente en parálisis fláccida arrefléxica, disfunción esfinteriana y con un nivel sensitivo. 7 pacientes (14,6%) padecieron síntomas durante menos de 1 mes, principalmente dolor y afectación sensitiva, con menor clínica motora. 22 pacientes (45,8%) mostraron sintomatología en un período de 1 a 6 meses, que consistió en dolor y debilidad de carácter progresivo; finalmente en 11 pacientes (23%) la clínica se observó durante un período de más de 6 meses, con similar cuadro al subgrupo anterior.
La sintomatología predominante se recoge en el siguiente gráfico:

4. Diagnóstico
La prueba diagnóstica fundamental, una vez sospechado por la clínica y/o radiología convencional, fue la RM. Como prueba complementaria en los casos de afectación ósea se empleó la TAC.
5. Tratamiento
Todos los pacientes fueron intervenidos, realizándose extirpación total de la lesión en 23 casos, parcial en 22 casos y biopsia simple en 3 pacientes. El abordaje más frecuentemente utilizado fue la vía posterior, con laminotomía/ laminoplastia de Raimondi. Se empleó microscopio quirúrgico y aspirador ultrasónico en algunos casos, sobre todo de neoplasias intramedulares. Se utilizaron abordaje de fosa posterior y cervical alto para las lesiones bulbares. Se emplearon abordajes combinados, en dos tiempos, con cirugía torácica o abdominal en neuroblastomas con extensión intrarraquídea. Se realizó tratamiento complementario con quimioterapia y/o radioterapia en algunos casos, como en neuroblastomas (quimioterapia), sarcoma de Ewing (quimioterapia + radioterapia), astrocitomas anaplásicos (radioterapia)...
6. Evolución postquirúrgica
Experimentaron mejoría 21 pacientes, sobre todo los que tenían lesiones extramedulares. Tuvieron una mejoría parcial dos niños con astrocitomas cervicales, uno con hemangioblastoma y otro con ganglioglioma dorsal. Se apreció déficit neurológico añadido o empeoramiento del previo en 6 casos, todos con astrocitomas intramedulares: un caso de glioblastoma cervical presentó tetraparesia e insuficiencia respiratoria que precisó traqueostomía; un caso de astrocitoma de cono medular sufrió trastorno esfinteriano definitivo; un caso de astrocitoma con extensión total de la médula desarrolló un síndrome medular completo con nivel T7; el resto de casos presentaron déficits variables con mejoría posterior.
Se produjeron dos éxitus en el período postoperatorio, un niño con osteosarcoma Cl-C2 que sufrió parada cardiorrespiratoria en el postoperatorio inmediato y un paciente con un astrocitoma bulbar que presentó insuficiencia respiratoria grave, complicada con neumonía masiva.
En el período postquirúrgico la mayoría de pacientes manifestaron alteraciones de sensibilidad del tipo de las parestesias o hipoestesias, autolimitadas en el tiempo. Dos pacientes con fístula de LCR y uno con hidrocefalia tras exéresis de una lesión bulbomedular requirieron la colocación de una válvula ventrículo-peritoneal. Un paciente presentó síndrome de Horner tras toracotomía para extirpación de neuroblastoma.
7. Seguimiento
Se produjeron tres recurrencias susceptibles de reintervención: una recidiva de un quiste óseo aneurismático a los 6 meses de la intervención y dos ependimomas de cola de caballo que se manifestaron como síndrome agudo de cola de caballo y como dolor ciático a los 22 y 6 meses tras la cirugía respectivamente.
Se produjeron 9 exitus, 2 en el postoperatorio inmediato (ya comentados) y el resto en pacientes con lesiones de alto grado de malignidad (glioblastoma, astrocitoma anaplásico, sarcoma de Ewing, PNET...).
Discusión
Los tumores espinales en la infancia suponen una entidad poco frecuente y de difícil diagnóstico, dada la habitual inespecificidad de los síntomas y su presentación subaguda-crónica. Con una media de retraso diagnóstico de alrededor de 6 meses, la sintomatología predominante fue dolor osteomuscular, amiotrofias o deformidades espinales. Especial mención requiere un paciente con un quiste aracnoideo y compresión medular que debutó con pies zambos y displasia de caderas, llegando a ser intervenido de estos problemas (se descubrió la patología causal 3 años más tarde). Otro caso, que cursó con tortícolis, escoliosis y posturas anómalas, presentaba un astrocitoma bulbomedular y fue diagnosticado 11 años más tarde cuando presentó tetraparesia, disfagia y nivel sensitivo C1. Según la mayoría de autores y al igual que en nuestra serie, la clínica principal suele consistir en alteraciones de la marcha y dolor axial o irradiado3,7,10-12, especialmente dolor continuo que interfiere con la vida cotidiana, en ocasiones acompañado de deformidades o rigideces espinales. En compresiones medulares importantes se aprecian además amiotrofias, piramidalismo, arreflexia, etc. En otros casos, la manifestación clínica consiste en la aparición de una tumoración14,15 hidrocefalia16 (por liberación masiva de proteínas) o meningismo17 (rotura de quiste dermoide).
En nuestra serie, los casos con clínica aguda no corresponden a lesiones intramedulares (excepto un cavernoma), sino a hemorragias por angiomas vertebrales, neuroblastomas o sarcoma de Ewing y recidivas de ependimoma con síndrome de cola de caballo. La mayoría de ellos fueron intervenidos de modo urgente/preferente con importante mejoría o con recuperación total en todos los casos.
El tumor más frecuente de esta serie fue el astrocitoma, con un 80% de lesiones de bajo grado cifra similar a la de otras series13,18-20. La mayoría se extienden a lo largo de varios segmentos vertebrales, fundamentalmente dorsales, debutando con la ya citada clínica algésica y motora. Tres pacientes con astrocitoma cervical o cervico-dorsal debutaron con síndrome disautonómico, con diaforesis, palidez y frialdad de extremidades. La extirpación completa resulta difícil en todos los casos por su extensión y mala diferenciación del tejido medular sano. En la literatura la exéresis completa varía del 14% al 88% según series7,13,21,22, citando algunos autores la poca repercusión de la misma en el pronóstico y evolución22. En nuestra experiencia la mayoría de resecciones fueron subtotales limitándose en algún caso a la biopsia; el objetivo principal fue preservar o mejorar el estado neurológico previo, lo que implica una menor agresividad quirúrgica. Tres pacientes con tumores de bajo grado empeoraron con respecto a la situación previa a la cirugía, recuperando función dos de ellos y quedando una lesión completa T7 en el tercero (astrocitoma panmedular). Los pacientes con afectación bulbocervical mostraron graves trastornos respiratorios postquirúrgicos produciéndose un caso de éxitus, otro necesitó una traqueostomía permanente y un tercero presentó una recuperación sin secuelas. La norma durante el seguimiento ha sido la estabilidad clínicorradiológica. Así mismo ha habido 4 pacientes con progresión de cifoescoliosis que han requerido una ortesis correctora.
El segundo diagnóstico en frecuencia ha sido el neuroblastoma, que ocurrió en edades medias más bajas que en el resto y que tuvo un mejor pronóstico en general (mejor cuanto menor edad). El cuadro varió según la existencia de invasión medular o propagación hacia otras regiones. El abordaje fue posterior en la mayoría de casos y combinado en el resto. La norma fue la recuperación neurológica, incluyendo un caso de paresia crural. Se administró tratamiento adyuvante con quimioterapia + radioterapia y se produjeron tres exitus por mala respuesta a estos tratamientos.
El manejo de las deformidades espinales consistió en fijación en el primer acto quirúrgico en cuatro casos con gran afectación ósea, una corpectomía con fijación vía anterior y posterior en un cordoma, fijación posterior en los quistes óseos aneurismáticos (una reintervención por hundimiento anterior) y una fijación occipitocervial en un osteosarcoma C1-C2. El resto de pacientes no precisaron fijación en un primer momento, aunque más tarde se realizó en los tres casos de progresión de cifoescoliosis que no se corrigieron con ortesis.
Conclusiones
La patología raquídea infantil cursa de modo insidioso y poco específico en su comienzo y evolución inicial, predominando el dolor y los síntomas motores. Estos, junto. con deformidades del raquis, deben hacer sospechar este tipo de lesiones e iniciar un estudio exhaustivo, ya que el diagnóstico precoz influirá en el tratamiento y evolución. La prueba de elección es la resonancia magnética. El pilar fundamental de tratamiento es la cirugía, con tratamientos adyuvantes de quimio-radioterapia según los casos.
Bibliografía
1. Beer, S.J., Menezes, A.H.: Primary tumors of the spine in children. Natural history, management, and long-term follow-up. Spine, 1997; 22: 649-658; discussion 658-659. [ Links ]
2. Cinalli, G., Sainte-Rose, C., Lellouch-Tubiana, A., Sebag, G., Renier, D., Pierre-Kahn, A.: Hydrocephalus associated with intramedullary low-grade glioma. Illustrative cases and review of the literature. J Neurosurg. 1995; 83: 480-485. [ Links ]
3. Curtiss, Ph. Jr., Collins, W.F.: Spinal-cord tumor--a cause of progressive neurological changes in children with scoliosis. A report of three cases. J Bone Joint Surg Am. 1961 ;43-A: 517-522. [ Links ]
4. DeSousa, A.L., Kalsbeck, J.E., Mealey, J. Jr., Campbell, R.L., Hockey, A.: Intraspinal tumors in children. A review of 81 cases. J Neurosurg. 1979; 51: 437-445. [ Links ]
5. Di Lorenzo, N., Giuffre, R., Fortuna, A.: Primary spinal neoplasms in childhood: analysis of 1234 published cases (including 56 personal cases) by pathology, sex, age and site. Differences from the situation in adults. Neurochirurgia (Stuttg). 1982; 25: 153-164. [ Links ]
6. Hasuo, K., Uchino, A., Matsumoto, S., Fujii, K., Fukui, M., Masuda, K.: MR imaging compared with CT, angiography, and myelography supplemented with CT in the diagnosis of spinal tumors. Radiat Med. 1993; 11: 177-186. [ Links ]
7. Innocenzi, G., Raco, A., Cantore, G., Raimondi, A.J.: Intramedullary astrocytomas and ependymomas in the pediatric age group: a retrospective study. Childs Nery Syst. 1996; 12: 776-780. [ Links ]
8. Jallo, G.I., Freed, D., Epstein, F.: Intramedullary spinal cord tumors in children. Childs Nery Syst. 2003; 19: 641-649. Epub 2003 Aug 8. [ Links ]
9. López-Ibor, B.: Neuroblastoma. En: Sierrasesúmaga, L. Oncología pedriática. Ed. Interamericana. McGraw Hill, 1992. [ Links ]
10. Lunardi, P., Missori, P.: Cranial and spinal tumors with meningitic onset. Ital J Neurol Sci. 1990; 11: 145-151. [ Links ]
11. Mehlman, C.T., Crawford, A.H., McMath, J.A.: Pediatric vertebral and spinal cord tumors: a retrospective study of musculoskeletal aspects of presentation, treatment, and complications. Orthopedics. 1999; 22: 49-55; discussion 55-56. [ Links ]
12. Menezes, A.H., Sato, Y.: Pediatr. Primary tumors of the spine in children--natural history and management. 1990. Neurosurgery. 1995; 23: 101-113; discussion 114. [ Links ]
13. Merchant, T.E., Kiehna, E.N., Thompson, S.J., Heideman, R., Sanford, R.A., Kun, L.E.: Pediatric low-grade and ependymal spinal cord tumors. Pediatr Neurosurg. 2000; 32: 30-36. [ Links ]
14. Merchant, T.E., Nguyen, D., Thompson, S.J., Reardon, D.A., Kun, L.E., Sanford, R.A.: High-grade pediatric spinal cord tumors. Pediatr Neurosurg. 1999; 30: 1-5. [ Links ]
15. Nadkarni, T.D., Rekate, H.L.: Pediatric intramedullary spinal cord tumors. Critical review of the literature. Childs Nery Syst. 1999; 15: 17-28. [ Links ]
16. O'Sullivan, C., Jenkin, R.D., Doherty, M.A., Hoffman, H.J., Greenberg, M.L.: Spinal cord tumors in children: long-term results of combined surgical and radiation treatment. J Neurosurg. 1994; 81: 507-512. [ Links ]
17. Parker, A.P., Robinson, R.O., Bullock, P.: Difficulties in diagnosing intrinsic spinal cord tumours. Arch Dis Child. 1996; 75: 204-207. [ Links ]
18. Payne, W.K. 3rd, Ogilvie, J.W.: Back pain in children and adolescents. Pediatr Clin North Am. 1996; 43: 899-917. [ Links ]
19. Pérez-Martínez, D.A., Bueno, H.J., Gutiérrez, F., López-Laso, E., Puente-Muñoz, A.l., Esparza, J., Simón, R., Mateos, F.: Spinal tumors in infancy. A report of 48 cases. Rev Neurol. 1999; 28: 863-867. [ Links ]
20. Ramos, R.: Papel del pediatra ante los tumores del sistema nervioso simpático. An. Esp. Pediatr 2000, 52: 261- 271. [ Links ]
21. Reimer, R., Onofrio, B.M.: Astrocytomas of the spinal cord in children and adolescents. J Neurosurg. 1985; 63: 669-675. [ Links ]
22. Schick, U., Marquardt, G.: Pediatric spinal tumors. Pediatr Neurosurg. 2001; 35: 120-127. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
José María Belinchón de Diego.
Hospital General Universitario de Valencia.
Servicio de Neurocirugía.
AV. Tres Cruces sn. 46014 Valencia.
Recibido: 17-08-07.
Aceptado: 2-05-08

