










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNeurocirugía
versión impresa ISSN 1130-1473
Neurocirugía vol.20 no.3 jun. 2009
Calpain inhibitor AK 295 inhibits calpain-induced apoptosis and improves neurologic function after traumatic spinal cord injury in rats
El inhibidor de la calpaina AK 295 inhibe la apoptosis inducida por calpaina y mejora la función neurológica tras traumatismo medular en ratas
A. Colak; M. Kaya*; A. Karaoglan; A. Sagmanligil*; O. Akdemir*; E. Sahan** and O. Celik***
Department of Neurosurgery, Maltepe University, School of Medicine, Istanbul; Departments of Neurosurgery* and Pathology** Taksim Education and Research Hospital, Taksim, Istanbul, Turkey. Department of Molecular Biology and Genetics***, Istanbul Kultur University Faculty of Science and Letters, Istanbul, Turkey.
SUMMARY
Background. An increase in the level of intracellular calcium activates the calcium-dependent neutral pro-tease calpain, which in turn leads to cellular dysfunction and cell death after an insult to the central nervous system. In this study, we evaluated the effect of a calpain inhibitor, AK 295, on spinal cord structure, neurologic function, and apoptosis after spinal cord injury (SCI) in a murine model.
Methods. Thirty albino Wistar rats were divided into 3 groups of 10 each: the sham-operated control group (group 1), the spinal cord trauma group (group 2), and the spinal cord trauma plus AK 295 treatment group (group 3). After having received a combination of ketamine 60 mg/kg and xylazine 9 mg/kg to induce anesthesia, the rats in groups 2 and 3 were subjected to thoracic trauma by the weight drop technique (40 g-cm). One hour after having been subjected to that trauma, the rats in groups 2 and 3 were treated with an intraperitoneal injection of either dimethyl sulfoxide 2 mg/kg or AK 295 2 mg/kg. The effects of the injury and the efficacy of AK 295 were determined by an assessment of the TUNEL technique and the results of examination with a light microscope. The neurologic performance of 5 rats from group 2 and 5 from group 3 was assessed by means of the inclined plane technique and the modified Tarlov's motor grading scale 1, 3, and 5 days after spinal cord trauma.
Findings. Light-microscopic examination of spinal cord specimens from group 2 revealed hemorrhage, edema, necrosis, and vascular thrombi 24 hours after trauma. Similar (but less prominent) features were seen in specimens obtained from group 3 rats. Twenty-four hours after injury, the mean apoptotic cell numbers in groups 1 and 2 were zero and 4.57 ± 0.37 cells, respectively. In group 3, the mean apoptotic cell number was2.30 ± 0.34 cells, a value significantly lower than that in group 2 (P < .05). Five days after trauma, the injured rats in group 2 demonstrated significant motor dysfunction (P < .05). In comparison, the motor scores exhibited by group 3 rats were markedly better (P < .05).
Conclusions. AK 295 inhibited apoptosis via calpaindependent pathways and provided neuroprotection and improved neurologic function in a rat model of SCI. To our knowledge, this is the first study to evaluate the use of AK 295, a calpain inhibitor, after SCI. Our data suggest that AK 295 might be a novel therapeutic compound for the neuroprotection of tissue and the recovery of function in patients with a SCI.
Key words: AK 295. Apoptosis. Calpain inhibitor. Secondary damage. Spinal cord trauma.
RESUMEN
Introducción. Una lesión en el sistema nervioso central origina un incremento en los niveles de calcio intracelular que activa la proteasa neutral calcio-dependiente calpaina, que a su vez conduce a la producción de disfunción y muerte celular. En este estudio evaluamos el efecto de un inhibidor de la calpaina, AK 295, sobre la estructura de la médula espinal, la función neurológica y apoptosis tras lesión medular en un modelo murino.
Métodos. Treinta ratas Wistar se dividieron en tres grupos de 10 ratas cada uno: Un grupo control (grupo 1), un grupo sometido a trauma espinal (grupo 2) y un grupo de ratas a las que se sometió a trauma medular y tratamiento con AK 295 (grupo 3). Después de recibir una combinación de ketamina 60mg/kg y xylazina 8mg/kg para la inducción anestésica, las ratas del grupo 2 y 3 fueron sometidas a trauma medular torácico mediante la técnica de caída de peso (40 g-cm). Una hora después de haber sufrido el traumatismo, las ratas del grupo 2 y 3 fueron tratadas mediante una inyección intraperitoneal bien de dimetil-sulfóxido 2mg/kg o de AK 295 2 mg/kg. Los efectos del traumatismo y la eficacia de AK 295 fueron determinados mediante la estimación de la técnica TUNEL y los resultados del examen del tejido mediante microscopía óptica. La función neurológica de 5 ratas del grupo 2 y 5 del grupo 3 fue estimada mediante la técnica del plano inclinado y la escala motora de Tarlov modificada a 1, 3 y 5 días desde el traumatismo medular.
Resultados. El estudio mediante microscopía óptica de las preparaciones de médula espinal del grupo 2 demostró la existencia de hemorragia, edema, necrosis y trombosis vascular 24 horas tras el traumatismo. Hallazgos similares pero menos importantes se encontraron en las preparaciones procedentes del grupo 3. Veinticuatro horas tras el trauma, el número medio de células apoptóticas en los grupos 1 y 2 fueron cero y 4.57 ± 0.37 células respectivamente. En el grupo 3, el número medio de células apoptóticas fue de 2.30 ± 0.34 células, un valor significativamente menor que en el grupo 2 (p < 0.05). Cinco días tras el traumatismo, las ratas lesionadas en el grupo 2 demostraron una significativamente mayor disfunción neurológica (p<0.05). En comparación, la puntuación motora que exhibieron las ratas del grupo 3 fue marcadamente mejor (p < 0.05).
Conclusión. AK 295 inhibe la apoptosis a través de vías calpain-dependientes y provee neuroprotección y consigue una mejor función neurológica en el modelo de lesión medular traumática en la rata. En nuestro conocimiento, este es el primer estudio en evaluar el uso de AK 295, un inhibidor de la calpaina, tras lesión medular traumática. Nuestros datos sugieren que AK 295 podría ser un nuevo compuesto terapéutico capaz de ofrecer neuroprotección tisular y recuperación funcional en pacientes con lesión medular traumática.
Palabras clave: AK 295. Apoptosis. Inhibidor de la calpaina. Lesión secundaria. Lesión medular traumática.
Introduction
Any type of trauma to the spinal cord causes significant primary and secondary damage. Secondary damage has been shown to induce the excessive release of excitatory amino acids and to cause intracellular calcium overload, the induction of free-radical-induced lipid peroxidation, and apoptosis (programmed cell death)24,47. Apoptosis is an actively regulated response that occurs after various cells have been subjected to external or internal stimuli. Recent experimental studies and clinical observations have revealed that spinal cord lesions are greatly exacerbated by apo ptosis10,12,13,15,18,19,22-24. However, the molecular and cellular mechanism of apoptosis is still not fully understood. The involvement of the caspases in apoptotic cell death does not exclude a role for other families of proteases such as calpains49. Both caspases and calpains are members of a group of proteases; the former is a calcium-independent protease, and the latter is a calcium-dependent protease. Laboratory findings and clinical evidence have suggested that caspases and calpains have an important role in the progression of programmed cell death and that multiple molecular pathways can lead independently to that type of cell death15,22-24,32 The activation of 1 protease can lead to the cleavage and activation of additional enzymes of the same or other proteases, and an amplified protease cascade results46.
In the CNS, calpain (which was discovered by Guroff) exists as an inactive pro-enzyme in the cytosol of resting cells16. It regulates the cytoskeleton, signal transduction, and metabolic pathways of healthy cells, and calcium is an essential ion for the activation of these cysteine proteases3,16. Increased calpain activation has been implicated not only in SCI but also in CNS-degenerative processes including Alzheimer's disease, Parkinson's disease, cerebral ischemia, and traumatic brain injury5,27,37,38,48. Blomgren and colleagues9 investigated the synergistic activation of caspase 3 by m-calpain after neonatal cerebral hypoxiaischemia in rats and suggested the presence of a direct link between the early calcium-mediated activation of calpain and the subsequent activation of caspase-3 that leads to a tentative pathway of pathologic apoptosis. Banik and colleagues3 introduced the calpain theory of CNS tissue destruction that occurred in rat spinal cord after impact injury. In a rat SCI model, Ray and colleagues33 demonstrated an increased expression and activity in the calpain that is involved in apoptosis. The increase in intracellular free Ca+2 that occurs immediately after an SCI triggers the upregulation of cysteine proteases and activates calpains2,17,28,51. Schumacher and colleagues39 demonstrated that significant calpain activation occurs within 15 minutes after experimentally induced SCI. Calpain activation also leads to the upstream of the caspase-3 that is involved in apoptotic cell death after SCI in rats32. Nakagawa and colleagues26described a new mode of caspase activation in which calpain is required for caspase-12 activation. They suggested that this novel pathway of calpain-mediated activation is induced by a disturbance in the intracellular calcium concentration. Calpain-mediated apoptosis occurs at least 4 to 6 hours after injury.
Usually, calpains are regulated by an endogenous calpain-specific inhibitor known as calpastatin. However, in pathologic conditions such as SCI, the targeting of the calpain (with or without caspase) cascade to prevent apoptosis remains a potential therapeutic strategy. Researcher has shown that blocking apoptosis with inhibitors of calpains or caspases improves neurologic outcome in subjects with an SCI7,12,19,31,32,34. Ray and colleagues30investigated the therapeutic efficacy of E-64-d, a calpain inhibitor, in SCI in a rat model. Those authors showed that the administration of the calpain inhibitor prevented apoptosis and restored the transcription of the genes for proteolipid protein and myelin basic protein. They concluded that E-64-d may provide functional protection in patients with an SCI. The anti-inflammatory agent methylprednisolone inhibits calpain and provides effective neuroprotection against calpain-mediated apoptosis in rats with an SCI4,34.
AK 295, which is considered to be an antiapoptotic agent that counteracts the effects of calpains, has been reported to have a role in the treatment of a variety of pathologic CNS conditions (including trauma and brain ischemia) induced in an experimental setting6,36. They showed that AK 295 provided significant neuroprotection of brain tissue and attenuated motor and cognitive deficits. The purpose of our study was to evaluate the effect of AK 295 on apoptosis and the recovery of motor function in a well-known rat model of SCI. To our knowledge, this is the first use of AK 295 after SCI.
Materials and methods
Preparation of animal subjects
Thirty albino Wistar rats (weight, approximately 250300 g) were used throughout the study. The rats were anesthetized with a combination of ketamine 60 mg/kg and xylazine 9 mg/kg. A longitudinal incision was made on the midline of the back, and the paravertebral muscles were dissected to expose vertebrae T8 to T10. A 3-level (T8-T10) laminectomy was performed to expose the spinal cord with the dura matter intact. Spinal cord injury was produced in the rats in groups 2 and 3 as described below. The layers were closed with 3/0 silk. During the surgical procedure and recovery from anesthesia, the rats were heated with a pad and a lamp to maintain their body temperature at 37ºC. They were then returned to their colony cage.
Experimental protocol
The rats were randomly divided into the following 3 groups of 10 each:
Group 1 (The sham-operated group). In each rat, a skin incision was made, and paravertebral muscle dissection, laminectomy, and closure of the anatomic layers were performed as described above. Twenty-four hours after surgery, 5 animals were reanesthetized as stated previously. The chest of each rat was opened, and via the intracardiac route, perfusion with 200 mL of serum physioloque was performed, after which the rats was fixed with 200 mL of neutral formaldehyde 10% solution according to the standard protocol. The dorsal region of each rat was reopened. The epicenter and 3 mm of tissue from the adjacent (rostral and caudal) regions of the spinal cord (a total about of 2 cm) were excised and harvested for examination via light microscopy and TUNEL staining. The remaining 5 animals were assessed 24 hours, 3 days, and 5 days after that procedure. At the end of 5 days, these 5 rats were reanesthetized and underwent perfusion and fixation as described before. Their spinal cord segments were removed and harvested for examination via light microscopy and TUNEL staining.
Group 2 (The trauma group). Laminectomies in group 2 rats were performed those in group 1 animals. Each rat was subjected to the thoracic trauma described below and was treated with 2 mg/kg dimethyl sulfoxide (DMSO). Five rats were killed 24 hours later, and their spinal cord samples were collected and processed as described above for group 1 animals. The remaining 5 rats were assessed for neurologic performance and were then killed, after which harvesting was performed 5 days after injury, as in group 1.
Group 3 (The trauma plus AK 295 treated group). Each rat in group 3 underwent laminectomy and was subjected to the spinal cord trauma described. AK 295 (Calbiochem GmBH; Med-Lab, Istanbul, Turkey) was dissolved in DMSO. One hour after trauma, the rats were treated with intraperitoneally administered AK 295 2 mg/kg, and that dose was repeated 24 hours after the trauma. The animals were sacrificed, and spinal cord samples were removed and prepared as stated above. Functional recovery assessments were performed as described in the other groups. Then the rats were killed and harvesting was performed in the manner described in groups 1 and 2.
Spinal cord injury model
SCI was induced via the drop-weight technique of Allen, which has been described previously in detail1. After thoracic laminectomy had been performed, a trauma of 40 g-cm was applied to each subject's spinal cord when a 4-g weight was dropped from a 10-cm height through a tube onto the exposed spinal cord.
Neurologic assessment
The neurologic status of animals was scored by assessing hind-limb neurologic function 24 hours, 3 days, and 5 days after SCI. Hind-limb function was assessed via the inclined plane technique of Rivlin and Tator35 and with the following modified version of Tarlov's grading scale45: grade 1, no voluntary hind-limb movement; grade 2, minimal movement but no ability to stand; grade 3, the ability to stand but the inability to walk; grade 4, the ability to walk with mild spasticity or incoordination of the hind limbs; and 5, the ability to walk normally.
Histopathologic examination
The spinal cord specimens were obtained both 24 hours and 5 days after trauma. The samples were fixed in a 10% neutral formaldehyde solution and were embedded in paraffin. Three serial sections were taken from the epicenter of the injury site and from an area 3 mm rostral to and 3 mm caudal to the epicenter. Tissue sections (thickness, 5µ) were cut transversely and were stained with hematoxylin and eosin before analysis was performed by means of a light microscope.
Detection of apoptotic cells
To detect apoptosis, we used a Frag ELTM DNA Fragmentation Colorimetric Detection Kit (Oncogene Research Products, San Diego, California, USA). This kit enables the identification of apoptotic nuclei in paraffin-embedded tissue sections via the end-labeling (FragEL) of DNA. To compare the extent of apoptosis after SCI in groups 2 and 3, all apoptotic cells were counted on TUNEL-stained sections from each of those 2 groups (n = 5). Sections were obtained from the epicenter (the impact site) and from 3 mm rostral to and 3 mm caudal to that area both 24 hours and 5 days after SCI. Cell counting was performed in 15 fields (5 rostral to the injury, 5 caudal to the injury, and 5 at the epicenter of the injury) per rat on the TUNEL-stained sections from each of those groups A blinded researcher counted all TUNEL-positive cells in these 15 fields, and then calculated an average count per field for each animal without discriminating between the white and the gray matter under high-power fields (original magnification x40) via light microscopy. The mean count per field for each set of specimens (24 hours and 5 days) in each group was calculated and compared.
Statistical analysis
The results of quantitative studies were expressed as the mean ± SD. Statistical comparisons between the groups were performed by means of the Kruskal-Wallis test and the Mann-Whitney U test. A P of < .05 was considered significant.
Results
Histopathologic examination
When examined with a light microscope, the spinal cord tissue from group 1 both 24 hours and 5 days after surgery revealed findings within normal limits, as expected. The group 2 tissue obtained 24 hours after trauma revealed diffuse hemorrhage, widespread edema, and focal necrosis in both gray-matter and white-matter regions, (especially in the gray-matter regions; Figure 1A). The severity of the trauma was prominent at that time. Significant damage was noted, and edema and hemorrhage in the dorsal surface of the spinal cord at the perilesional level were evident. A significant loss of myelin and axons was also observed in the white matter. The specimens obtained 5 days after trauma showed cystic vacuolar degeneration, vascular thrombi, and dispersion of the myelin sheaths. Edema and an accumulation of fibrin were also seen throughout the spinal cord. The traumatized spinal cord was infiltrated with polymorphonuclear leucocytes, erythrocytes, and macrophages (Figure 1B).

Figure 1. Trauma-exposed rats; group 2:
A) A photomicrograph of the white matter 24 hours
after injury reveals widespread hemorrhage and
edema with focal fibrinoid necrosis (hematoxylin-eosin
stain, original magnification x40). B) A photograph
of the gray-white mater junction region 5 days after
injury shows severe vacuolar cystic defects and nerve
fiber necrosis. Note the hemorrhagic and edematous
areas with an extensive infiltration of polymorphonuclear
leucocytes and macrophages (hematoxylineosin stain,
original magnification x10).
Twenty-four hours after injury, the architecture of the spinal cord in group 3 was better preserved than that in group 2. In the spinal cord specimens from group 3, there were moderate hemorrhagic changes, not only in the gray matter but also in the white-matter region (Figure 2A). In contrast to the findings in group 2, edema and focal necrosis were less prominent. On the fifth day after injury in group 3, the cellular structures of the spinal cord were also better preserved than those in group 2 rats (Figure 2B). There was no widespread edema, but small areas of mild edema were visible on some slides. Neither large vacuoles nor vascular thrombi were observed. In some areas of the white or gray matter, a few polymorphonuclear leucocytes and macrophages were identified. Those findings suggested that AK 295 treatment significantly improved the histopathologic changes after SCI.

Figure 2, A and B. AK 295-treated rats; group 3.
A) A photograph of the gray matter 24 hours after
trauma. Note the multifocal petechial hemorrhage
and the mild edema with focal necrosis in some areas
(hematoxylin-eosin stain, original magnification x40).
B) Examination via a light microscope of the gray-white
matter 5 days after trauma reveals mild cystic vacuolar
degeneration and hemorrhage (hematoxylin-eosin stain,
original magnification x40).
Apoptotic cell count
Almost no apoptotic cells were detected in the spinal cord of group 1 rats (Figure 3A). After 24 hours and after 5 days, the tissues obtained from group 2 rats contained dark brown apoptotic cells and intercellular apoptotic fragments in both the gray and the white matter of the spinal cord tissue (Figure 3B). In contrast, specimens obtained from group 3 rats contained a small number of apoptotic cell or fragments (Figure 3C). The mean apoptotic cell number in group 2 rats 24 hours after injury was 4.57 ± 0.37 cells, and that in group 3 rats was 2.30 ± 0.34 cells (Figure 4). The mean apoptotic cell numbers 5 days after injury are shown in Figure 4.

Figure 3, A-C. A) There were no apoptotic cells in
the spinal cord specimens taken from sham-operated
rats (TUNEL, original magnification x40). B) The number
of apoptotic cell (arrows) increased 24 hours after SCI
(TUNEL, original magnification x40). C) In contrast, only
a few apoptotic cells (arrows) were noted in the spinal
cord specimens obtained from AK 295-treated rats (TUNEL,
original magnification x40).
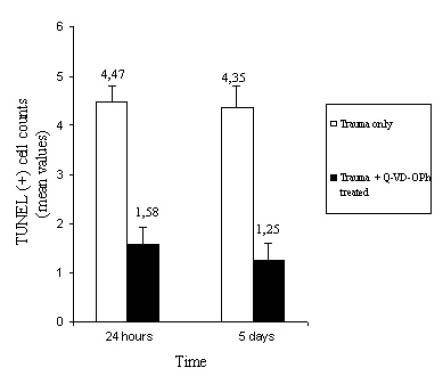
Figure 4. A graph showing the apoptotic cell count. After injury,
an increase in the apoptotic cell number in the spinal cord samples
from group 2 rats was noted both 24 hours and 5 days after injury.
Treatment with AK 295 significantly reduced the SCI-induced increase
in the number of apoptotic cells at both time points (P < .05). The data
are the mean ± SD.
Neurologic performance
Twenty-four hours after injury, the mean inclined plane degrees of the animals were 65.0 ± 1.76 in group 1, 42.00 ± 2.09 in group 2, and 46.50 ± 2.23 in group 3. On the fifth day after injury, those values were 65.0 ± 1.76, 42.50 ± 1.36, and 50.50 ± 2.70, respectively (Figure 5A). The differences between group 2 and 3 rats were significant in all time periods after injury (P < .05). Twenty-four hours after injury, the mean motor grading scores were 5.0 ± 0.0 in group 1, 1.0 ± 0.0 in group 2, and 1.20 ± 0.24 in group 3. The mean motor grading scores after injury are shown in Figure 5B. The differences in those scores between groups 2 and 3 were significant (P < .05).
Discussion
Our findings indicate that AK 295 produces a beneficial effect after SCI in a murine model. We noted that the limitation of injury and ultimate recovery differed significantly in treated rats as opposed to untreated rats. The neuroprotective effect of AK 295 involves the inhibition of calpain and related caspases as well as the stabilization of the cal-pain-caspase system that is altered after SCI. Calpain activation, which is an early feature of injury after SCI, occurs within 15 minutes after the insult, peaks 2 hours later, and decreases to a value within normal limits 16 to 24 hours after injury39. For that reason in our experiment, calpain inhibitor was administered immediately after injury, and its efficacy was investigated histopathologically both 24 hours and 5 days after trauma.
A primary injury to the spinal cord causes both morphologic and biochemical changes that initiate secondary pathophysiologic pathways and destroy CNS cells and nerve fibers. In living cells, cytoskeletal and membrane proteins maintain the structural integrity of cells29. Several mammalian calpains have been identified, most of which have been shown to contribute cell death8,21,43. An increase in the intracellular free Ca+2 concentration triggers the calpain activation that cleaves many cytoskeletal and myelin proteins, which eventually results in apoptosis2,17,28,51. The overactivation of calpain is involved in the pathophysiology of neurodegenerative disorders and diseases and increases the lesion size as time passes after SCI5,27,37,38,48. After a primary injury to the spinal cord, the process of secondary injury progresses over time in the both rostral and caudal areas; this increases the lesion size and the rate of cell death. Research has shown that calpain is involved in the mediation of apoptotic cell death, not only in SCI lesions but also in regions distant from the initial site of injury31,32,34. Similar results were observed in this experiment and in other studies of caspases that we performed in our laboratory and that were conducted by other researchers7,10-12.
Research has revealed that an SCI evokes an increase in the intracellular free Ca+2 level, which in turn results in the activation of calpain17,51. Ray and colleagues29 demonstrated an increased calpain expression and activity that are responsible for neuronal apoptosis after SCI in the rat. Other researchers have suggested that Ca+2 accumulation and calpain activation are associated with the caspase cascade leading to apoptosis after SCI14,17,31,32,39,51. Increased calpain expression has been detected in various cells in SCI lesions as well as in the penumbra after an SCI33,41. Ray and colleagues32 investigated the relationship between calpain and caspase-3 in a rat model and clarified the role of both cysteine proteases in the pathophysiologic mechanism of SCI. They concluded that calpain works upstream of caspase in the mediation of apoptosis after SCI. Similar observations were noted in a pathologic apoptosis study of neonatal cerebral hypoxia-ischemia in rats, which was conducted by Blomgren and colleagues9. Those authors suggested the existence of a direct link between the early calciummediated calpain activation and the subsequent caspase-3 activation. Nakagawa and colleagues26 showed a novel ERspecific pathway mediated by caspase-12. They revealed that calpain activation correlates with caspase-12 cleavage activity that is evoked by the mobilization of the intracellular calcium store. The association between the calpain activity and caspase-12 cleavage was demonstrated in an experimental study performed by Wootz and colleagues on the spinal cord of transgenic mice with amyotrophic lateral sclerosis50. In our study, the apoptotic cell number in group 2 rats 24 hours after injury was significantly increased after trauma to the spinal cord, and in group 3 rats, that number was significantly decreased after treatment with AK 295 (4.57 ± 0.37 cells in group 2 versus 2.30 ± 0.34 cells in group 3) (P < .05; Figure 4). Apoptotic cell death was noted in neurons and glia, not only in the gray matter but also in the white matter of the injured spinal cord (Figures 1 and 3B). We suggest that trauma to the spinal cord caused an activation of caspases and an increase in the intracellular calcium concentration that triggered calpain activation. Both cysteine proteases then cleaved to each other via the caspase-3 and caspase-12 pathways, and the result was significant apoptotic cell death.
A therapeutic strategy using inhibitors of calpain appears to be effective in the neuroprotection and functional recovery of CNS tissues. Calpastatin is a specific endogenous inhibitor that regulates the proteolytic activity of calpains in mammalian cells25,42,44. It is a very large molecule that is not cell permeable. Therefore, it is not a well target in an anti-calpain treatment. However, calpain inhibitors should be cell permeable and should also have a higher specificity. Schumacher and colleagues40 investigated the efficacy of CEP-4143, a μ-calpain inhibitor, in an animal model of experimentally induced SCI. Those authors demonstrated for the first time that this agent significantly preserved tissue and improved the behavioral outcome of the study subjects. AK 295 is a tripeptidyl α-keto amide that strongly inhibits both forms (μ and m) of calpain-reversible. It is lipophilic and is also a selective inhibitor of calpain. Bartus and colleagues6, who investigated the effect of AK 295 in a model of focal ischemic brain damage, showed that the intra-arterial administration of that molecule protected neurons from ischemia. Saatman and colleagues36 investigated the efficacy of AK 295 in a murine model of experimentally induced traumatic injury. Those investigators showed that calpain has a role in posttraumatic events. They also showed that the postinjury administration of AK 295 improved motor and cognitive deficits measured 1 week after injury in the study animals. To our knowledge, our study is the first to evaluate the effects of AK 295 in treating SCI. We concluded that AK 295 saved neurons and maintained their axon-myelin structural unit by preventing the degradation of cytoskeletal and membrane proteins and therefore preserved motor function as demonstrated via assessment with light microscopy and neurologic testing (Figures 2, 3C, 5).
Conclusion
In our study, the administration of 2 mg/kg of AK 295 after SCI inhibited the activity of calpain and its related caspases. A decrease in apoptotic cell death and lesion size resulted, and motor function was maintained because axon-myelin integrity had been preserved. AK 295, which has been shown to inhibit calpain activity and aid tissue recovery, is a promising agent for use in the neuroprotective treatment of acute SCI in humans. However, further experimental and clinical studies are needed to determine the exact protective mechanisms involved.
Acknowledgments
We thank Dr. Veysel Antar, Dr. Şeref Barut, and Dr. Bilal Kelten for their technical assistance and Dr. Aysenur Akyildiz for their help and comments.
References
1. Allen, A.R.: Remarks on the histopathological changes in the spinal cord due to impact. An experimental study. J Nerv Ment Dis 1914; 1: 141-147. [ Links ]
2. Banik, N.L., Hogan, E.L., Powers, J.M., Smith, K.P.: Proteolytic enzymes in experimental spinal cord injury. J Neurol Sci 1986; 73: 245-256. [ Links ]
3. Banik, N.L., Hogan, E.L., Powers, J.M., Whetstine, L.J.: Degradation of cytoskeletal proteins in experimental spinal cord injury. Neurochem Res 1982; 7: 1465-1475. [ Links ]
4. Banik, N.L., Matzelle, D., Terry, E., Hogan, E.L.: A new mechanism of methylprednisolone and other corticosteroids action demonstrated in vitro: inhibition of a proteinase (calpain) prevents myelin and cytoskeletal protein degradation. Brain Res 1997; 14: 205-210. [ Links ]
5. Banik, N.L., Shields, D.C., Ray, S., et al.: Role of calpain in spinal cord injury: effects of calpain and free radical inhibitors. Ann N Y Acad Sci 1998; 30: 131-137. [ Links ]
6. Bartus, R.T., Hayward, N.J., Elliott, P.J., et al.: Calpain inhibitor AK295 protects neurons from focal brain ischemia. Effects of postocclusion intra-arterial administration. Stroke 1994; 25: 2265-2270. [ Links ]
7. Barut, S., Unlu, Y.A., Karaoglan, A., et al.: The neuroprotective effects of z-DEVD.fmk, a caspase-3 inhibitor, on traumatic spinal cord injury in rats. Surg Neurol 2005; 64: 213-220. [ Links ]
8. Blanchard, H., Grochulski, P., Li, Y., et al.: Structure of a calpain Ca(2+)-binding domain reveals a novel EF-hand and Ca(2+)-induced conformational changes. Nat Struct Biol 1997; 4: 532-538. [ Links ]
9. Blomgren, K., Hallin, U., Andersson, A.L., et al.: Calpastatin is up-regulated in response to hypoxia and is a suicide substrate to calpain after neonatal cerebral hypoxia-ischemia. J Biol Chem 1999; 14: 14046-14052. [ Links ]
10. Bresnahan, J.C., Schuman, S.L., Beattie, M.S.: Evidence for apoptosis of oligodendroglia in long tracts undergoing wallerian degeneration after spinal cord injury (SCI) in monkeys. Soc Neurosci Abstr 1996; 22: 1185. [ Links ]
11. Casha, S., Yu, W.R., Fehlings, M.G.: Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience 2001; 103: 203-218. [ Links ]
12. Colak, A., Karaoglan, A., Barut, S., et al.: Neuroprotection and functional recovery after application of the caspase-9 inhibitor z-LEHD-fmk in a rat model of traumatic spinal cord injury. J Neurosurg Spine 2005; 2: 327-334. [ Links ]
13. Crowe, M.J., Bresnahan, J.C., Shuman, S.L., et al.: Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3: 73-76. Erratum in: Nat Med 1997, 3: 240. [ Links ]
14. Du, S., Rubin, A., Klepper, S., et al.: Calcium influx and activation of calpain I mediate acute reactive gliosis in injured spinal cord. Exp Neurol 1999; 157: 96-105. [ Links ]
15. Emery, E., Aldana, P., Bunge, M.B., et al.: Apoptosis after traumatic human spinal cord injury. J Neurosurg 1998; 89: 911-920. [ Links ]
16. Guroff, G.: A neutral, calcium-activated proteinase from the soluble fraction of rat brain. J Biol Chem 1964; 239: 149-155. [ Links ]
17. Happel, R.D., Smith, K.P., Banik, N.L., et al.: Ca2+accumulation in experimental spinal cord trauma. Brain Res 1981; 211: 476-479. [ Links ]
18. Katoh, K., Ikata, T., Katoh, S., et al.: Induction and its spread of apoptosis in rat spinal cord after mechanical trauma. Neurosci Lett 1996; 216: 9-12. [ Links ]
19. Keane, R.W., Kraydieh, S., Lotocki, G., et al.: Apoptotic and anti-apoptotic mechanisms following spinal cord injury. J Neuropathol Exp Neurol 2001; 60: 422-429. [ Links ]
20. Li, Z., Hogan, E.L., Banik, N.L.: Role of calpain in spinal cord injury: increased calpain immunoreactivity in rat spinal cord after impact trauma. Neurochem Res 1996; 21: 441-448. [ Links ]
21. Lin, G.D., Chattopadhyay, D., Maki, M., et al.: Crystal structure of calcium bound domain VI of calpain at 1.9 A resolution and its role in enzyme assembly, regulation, and inhibitor binding. Nat Struct Biol 1997; 4: 539-547. [ Links ]
22. Liu, X.Z., Xu, X.M., Hu, R., et al.: Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci 1997; 17: 5395-5406. [ Links ]
23. Lou, J., Lenke, L.G., Ludwig, F.J., O'Brien, M.F.: Apoptosis as a mechanism of neuronal cell death following acute experimental spinal cord injury. Spinal Cord 1998; 36: 683-690. [ Links ]
24. Lu, J., Ashwell, K.W., Waite, P.: Advances in secondary spinal cord injury: role of apoptosis. Spine 2000; 25: 1859-1866. [ Links ]
25. Mohan, P.S., Nixon, R.A.: Purification and properties of high molecular weight calpastatin from bovine brain. J Neurochem 1995; 64: 859-866. [ Links ]
26. Nakagawa, T., Zhu, H., Morishima, N., et al.: Caspase12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000; 403: 98-103. [ Links ]
27. Ray, S.K., Banik, N.L.: Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord 2003; 2: 173- 189. [ Links ]
28. Ray, S.K., Banik, N.L.: Calpain. Wiley Encycl Mol Med 2002; 5: 435-440. [ Links ]
29. Ray, S.K., Hogan, E.L., Banik, N.L.: Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res Brain Res Rev 2003; 42: 169-185. [ Links ]
30. Ray, S.K., Matzelle, D.D., Sribnick, E.A., et al.: Calpain inhibitor prevented apoptosis and maintained transcription of proteolipid protein and myelin basic protein genes in rat spinal cord injury. J Chem Neuroanat 2003; 26: 119-124. [ Links ]
31. Ray, S.K., Matzelle, D.D., Wilford, G.G., et al.: Increased calpain expression is associated with apoptosis in rat spinal cord injury: calpain inhibitor provides neuroprotection. Neurochem Res 2000; 25: 1191-1198. [ Links ]
32. Ray, S.K., Matzelle, D.D., Wilford, G.G., et al.: Inhibition of calpain-mediated apoptosis by E-64 d-reduced immediate early gene (IEG) expression and reactive astrogliosis in the lesion and penumbra following spinal cord injury in rats. Brain Res 2001; 916: 115-126. [ Links ]
33. Ray, S.K., Shields, D.C., Saido, T.C., et al.: Calpain activity and translational expression increased in spinal cord injury. Brain Res 1999; 816: 375-380. [ Links ]
34. Ray, S.K., Wilford, G.G., Matzelle, D.C., et al.: Calpeptin and methylprednisolone inhibit apoptosis in rat spinal cord injury. Ann N Y Acad Sci 1999; 890: 261-269. [ Links ]
35. Rivlin, A.S., Tator, C.H.: Objective clinical assessment of motor function after experimental spinal cord injury in the rat. J Neurosurg 1977; 47: 577-581. [ Links ]
36. Saatman, K.E., Murai, H., Bartus, R.T., et al.: Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci U S A 1996; 93: 3428-3433. [ Links ]
37. Saido, T.C., Kawashima, S., Tani, E., Yokota, M.: Up-and down-regulation of calpain inhibitor polypeptide, calpastatin, in postischemic hippocampus. Neurosci Lett 1997; 227: 75-78. [ Links ]
38. Saito, K., Elce, J.S., Hamos, J.E., Nixon, R.A.: Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A 1993; 90: 2628-2632. [ Links ]
39. Schumacher, P.A., Eubanks, J.H., Fehlings, M.G.: Increased calpain I-mediated proteolysis, and preferential loss of dephosphorylated NF200, following traumatic spinal cord injury. Neuroscience 1999; 91: 733-744. [ Links ]
40. Schumacher, P.A., Siman, R.G., Fehlings, M.G.: Pretreatment with calpain inhibitor CEP-4143 inhibits calpain I activation and cytoskeletal degradation, improves neurological function, and enhances axonal survival after traumatic spinal cord injury. J Neurochem 2000; 74: 1646-1655. [ Links ]
41. Shields, D.C., Schaecher, K.E., Hogan, E.L., Banik, N.L.: Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J Neurosci Res 2000; 61: 146-150. [ Links ]
42. Sorimachi, Y., Harada, K., Saido, T.C., et al.: Down-regulation of calpastatin in rat heart after brief ischemia and reperfusion. J Biochem (Tokyo) 1997; 122: 743-748. [ Links ]
43. Strobl, S., Fernandez-Catalan, C., Braun, M., et al.: The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc Natl Acad Sci U S A 2000; 97: 588-592. [ Links ]
44. Suzuki, K., Imajoh, S., Emori, Y., et al.: Calcium-activated neutral protease and its endogenous inhibitor. Activation at the cell membrane and biological function. FEBS Lett 1987; 220: 271-277. [ Links ]
45. Tarlov, I.M.: Spinal Cord Compression: Mechanism of Paralysis and Treatment. Charles C. Thomas, Springfield, Illinois, 1957; p: 147. [ Links ]
46. Tewari, M., Quan, L.T., O'Rourke, K., et al.: Yama/ CPP32 beta, a mammalian homolog of CED-3, is a CrmAinhibitable protease that cleaves the death substrate poly (ADP-ribose) polymerase. Cell 1995; 81: 801-809. [ Links ]
47. Thompson, C.B.: Apoptosis in the pathogenesis and treatment of disease. Science 1995; 267: 1456-1462. [ Links ]
48. Tsuji, T., Shimohama, S., Kimura, J., Shimizu, K.: m-Calpain (calcium-activated neutral proteinase) in Alzheimer's disease brains. Neurosci Lett 1998; 248: 109-112. [ Links ]
49. Villa, P.G., Henzel, W.J., Sensenbrenner, M., et al.: Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J Cell Sci 1998; 111: 713-722. [ Links ]
50. Wootz, H., Hansson, I., Korhonen, L., Lindholm, D.: XIAP decreases caspase-12 cleavage and calpain activity in spinal cord of ALS transgenic mice. Exp Cell Res 2006; 312: 1890-1898. [ Links ]
51. Young, W., Yen, V., Blight, A.: Extracellular calcium ionic activity in experimental spinal cord contusion. Brain Res 1982; 253: 105-113. [ Links ]
![]() Correspondence:
Correspondence:
Ahmet Colak, MD.
Terakki Caddesi, 47/7,
Kartaltepe, Bakirkoy. Istanbul, Turkey.
Received: 17-05-08.
Accepted: 4-01-09.

