










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkFarmacia Hospitalaria
On-line version ISSN 2171-8695Print version ISSN 1130-6343
Farm Hosp. vol.37 n.6 Toledo Nov./Dec. 2013
https://dx.doi.org/10.7399/FH.2013.37.6.872
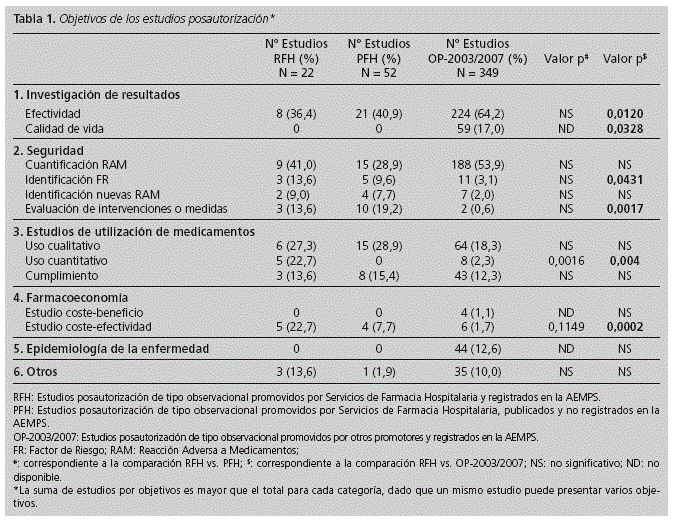
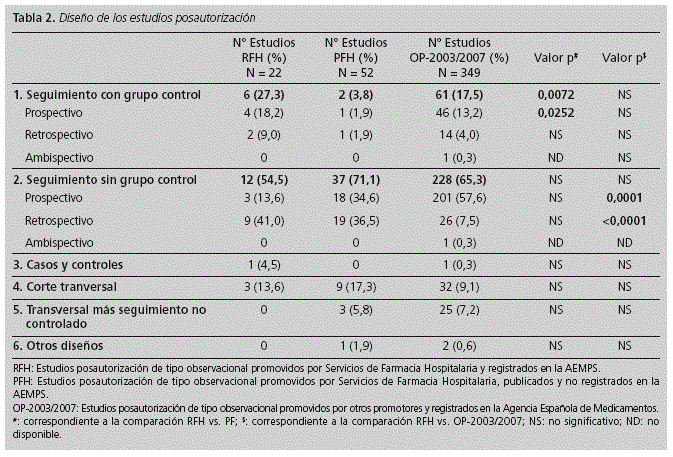
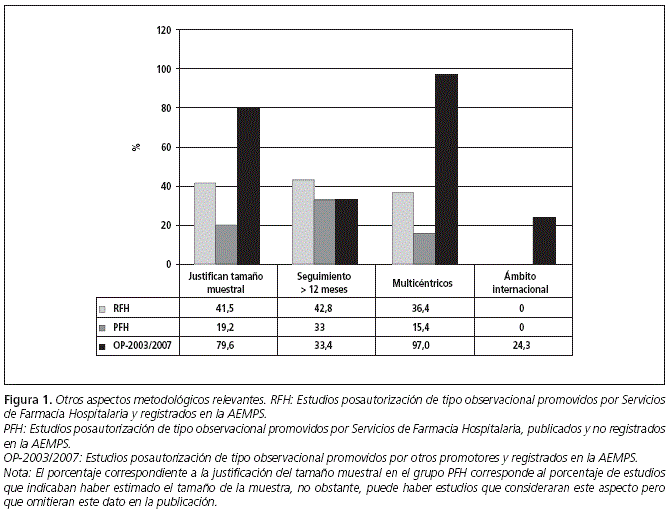
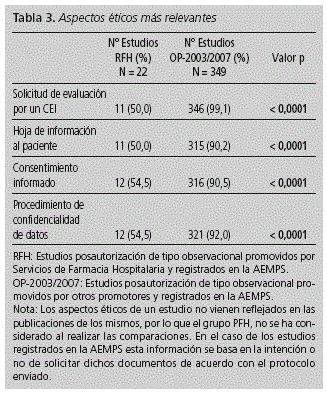
ORIGINALES Calidad ética y metodológica de los estudios posautorización de tipo observacional promovidos por Servicios de Farmacia Hospitalaria Ethical and methodological quality of non-interventional post-authorization studies promoted by hospital pharmacy departments D. González Bermejo1, M.a P. Vicente Sánchez2, C. Pozuelo González3, D. Macías Saint-Gerons1, V. Greciano Greciano1 y C. de la Fuente Honrubia1 1Agencia Española de Medicamentos y Productos Sanitarios. Ministerio de Sanidad, Servicios Sociales e igualdad. Madrid. Dirección para correspondencia RESUMEN Objetivos: Describir la calidad ética y metodológica de los estudios posautorización de tipo observacional con medicamentos de uso humano promovidos por los Servicios de Farmacia Hospitalaria (SFH). Palabras clave: Observacional; Posautorización; Postautorización; Investigación. ABSTRACT Objectives: To describe the ethical and methodological quality of non-interventional post-authorization studies promoted by Hospital Pharmacy Departments (HPD). Key words: Non-interventional; Post-authorization; Research. Introducción El farmacéutico de hospital, por su formación, acceso a fuentes de información sobre medicamentos y experiencia en el seguimiento farmacoterapéutico de los pacientes, puede aportar información relevante sobre la efectividad, seguridad y patrones de uso de los medicamentos cuando éstos son utilizados en condiciones reales, en aquellas áreas en las que se haya detectado falta de evidencia o para resolver dudas concretas derivadas de su uso1,2. En este sentido, la participación del farmacéutico en estudios de tipo observacional con medicamentos puede ser de gran utilidad. Publicaciones previas han descrito el papel del farmacéutico de hospital en los ensayos clínicos3,4 . No obstante, no se ha analizado su papel en los estudios observacionales. Cabe mencionar que las normativas que aplican a ambos tipos de estudios son igualmente de obligado cumplimiento, con independencia del promotor o responsable del proyecto (profesional sanitario, sociedad científica, fundación, universidad o industria farmacéutica)2,4 . Los estudios promovidos y realizados por farmacéuticos de hospital poseen un valor adicional al considerarse que recogen el uso real del medicamento y sus efectos sin factores de distorsión, al poder establecer los mecanismos que garanticen una disociación efectiva entre la prescripción de la medicación y la participación del paciente en el estudio6,7. Esto constituye una ventaja sobre los estudios en los que la investigación del uso de un medicamento corre a cargo de quien lo prescribe, pues es conocido que ello puede alterar los hábitos del prescriptor y conducir a sesgos en los resultados -Efecto Hawthorne8-. No obstante, para obtener resultados válidos, estos estudios han de contar con las garantías necesarias para asegurar una calidad óptima desde un punto de vista ético y metodológico, exigible a todo tipo de investigación clínica. Estos requisitos se recogen en la Orden SAS 3470/2009, con la que es preciso estar familiarizado a la hora de llevar a cabo estudios observacionales con medicamentos. Este artículo, pretende describir las características de los estudios observacionales promovidos por los Servicios de Farmacia Hospitalaria (SFH), su calidad ética y metodológica, así como su adecuación a la normativa aplicable. Materiales y métodos Se ha recogido información de todos aquellos estudios posautorización de tipo observacional (EPA) promovidos por los SFH y registrados (RFH) en la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) en el período comprendido entre el 25 de diciembre de 2009 (fecha en la que se publicó la Orden SAS 3470/20095) y 31 de diciembre de 2011. La información de los estudios registrados en la AEMPS se obtuvo de la base de datos GESTO (Gestión de Estudios Observacionales), en la que se incluyen datos administrativos, metodológicos, aspectos éticos, información de seguimiento y dictámenes emitidos por las autoridades sanitarias. De cada unos de los protocolos de los estudios se recogieron datos administrativos (promotor, monitor), datos metodológicos (ámbito de estudio, fuente de información, diseño del estudio, objetivos, número de centros, ámbito asistencial, notificación de reacciones adversas y período de estudio), datos de la medicación objeto de estudio (principio activo, grupo farmacológico), aspectos éticos (consentimiento informado, hoja de información al paciente, procedimiento de confidencialidad de datos, solicitud de evaluación a un Comité de Ética de la Investigación -CEI-), resolución de las autoridades competentes y tipo de estudio según clasificación asignada por la AEMPS, de acuerdo con la Orden SAS 3470/20095. Para caracterizar mejor la calidad de los estudios promovidos por los SFH, y poder contar con un grupo de estudios de referencia registrados en la AEMPS que pudiera ser comparable, se identificaron también los estudios posautorización de tipo observacional registrados en el período 2003-2007 realizados por "Otros Promotores" (OP-2003-2007), para los cuales existen datos disponibles y publicados por la AEMPS9. Con el fin de conocer la calidad ética y metodológica de aquellos EPA promovidos por SFH que pudieran no haber sido registrados por la AEMPS, se analizaron como muestra representativa los artículos originales y originales breves publicados en la revista Farmacia Hospitalaria entre los años 2009 y 2011 (PFH). Se recogieron, cuando estaban disponibles en la publicación, los parámetros mencionados anteriormente relativos a las características y calidad de los estudios. Para determinar si el cumplimiento con la normativa mejora la calidad de los EPA promovidos por SFH, se realizó una comparación estadística de aquellas variables que se consideraron relevantes, entre los RFH (cumplidores de la normativa) y los PFH no registrados en la AEMPS (no cumplidores de la normativa). Los resultados de las variables discretas se expresaron como porcentaje y las de las variables continuas como la mediana y amplitud intercuartil (percentil 25 [p25] y percentil 75 [p75]). Para la comparación de dos porcentajes se empleó la prueba exacta de Fisher. Las variables continuas se analizaron mediante la prueba U Whitney. Resultados En el período estudiado se registraron en la AEMPS 744 EPA, de los cuales 22 (3%) eran estudios RFH. La potencia estadística alcanzada para las comparaciones que incluían las variables "estimación del tamaño muestral" y "solicitud de evaluación por un CEI" fue superior al 95% (error alfa = 0,05). Estos cálculos están disponibles 24 mediante solicitud a los autores. Entre aquellos estudios cuyos promotores son investigadores independientes de unidades clínicas pertenecientes a hospitales y centros sanitarios (283), los estudios RFH suponen un 5%. El resto de promotores están representados por los siguientes sectores: industria farmacéutica: 328 (44%), sociedades científicas: 96 (13%), fundaciones e institutos de investigación: 30 (4%), universidades: 7 (1%). De los estudios RFH, 7 (32%) preveían un seguimiento prospectivo de los participantes y 15 (68%) presentaban otros diseños diferentes. Todos estos EPA fueron de ámbito nacional, 8 (36%) fueron multicéntricos y el historial clínico fue la fuente de información en gran parte de ellos (9 [41%]). En 15 estudios se proponía un tamaño muestral, previendo incluir un total de 785.210 pacientes (mediana = 300; p25 = 80; p75 = 700). Los objetivos generales más estudiados fueron seguridad, utilización de medicamentos e investigación de resultados (Tabla 1) y los grupos terapéuticos más investigados fueron los antineoplásicos e inmunomoduladores (36,4%) y los 12 antiinfecciosos (22,7%). El diseño de los estudios se presenta en la tabla 2. Entre los 18 EPA de tipo longitudinal (82%), 6 (33%) tenían grupo control. A su vez, de los 11 los estudios que presentaban como objetivo evaluar la seguridad de medicamentos 4 disponían de grupo control (36%), encontrando un porcentaje similar (3, [33%]) entre los 9 estudios que evaluaban investigación de resultados. El tamaño muestral y otros aspectos metodológicos se muestran en la figura 1. El ámbito en el que mayoritariamente se pretendían desarrollar estos estudios fue atención especializada (consultas) (50%), seguido de hospitalización (ingresos) (36%) y ámbito mixto (atención especializada y primaria, 14%). El plan de trabajo del estudio figuraba en el 55% de los protocolos, mientras que tan sólo 1 de los 7 estudios de seguimiento prospectivo, recogía el procedimiento de notificación de sospechas de reacciones adversas. En ninguno de estos últimos, se preveía en el protocolo la presentación de informes de seguimiento a las autoridades sanitarias y tampoco constaba la solicitud de autorización a los organismos competentes de las comunidades autónomas correspondientes. En un único estudio (4,5%) se preveía el envío de informe final a las autoridades competentes una vez concluido el estudio. Respecto a los aspectos éticos (Tabla 3), 11 estudios (50%) reflejaban la intención de solicitar la revisión por parte de un CEI o aportaban un documento que acreditara dicha revisión. El procedimiento de confidencialidad de datos del paciente se describía en 10 estudios (45%). Asimismo, se preveía solicitar consentimiento informado en 12 estudios (55%) y 11 (50%) presentaban hoja de información al paciente. Gran parte de los estudios que no presentaban procedimiento de confidencialidad de datos, hoja de información al paciente o no preveían la solicitud de consentimiento informado al paciente se correspondía con aquellos que no indicaron su intención de solicitar valoración a un CEI (70%, 64% y 70% respectivamente). Al comparar estos resultados con los datos disponibles para los estudios OP-2003/2007 (N = 349), se observaron ciertas diferencias. En cuanto a los objetivos de investigación, los estudios RFH investigaban en mayor medida de los factores de riesgo para desarrollar eventos adversos, evaluación de intervenciones o medidas, el uso cuantitativo de los medicamentos y aspectos farmacoeconómicos. Por el contrario, un mayor porcentaje de los registrados en 2003/2007, estudiaban la efectividad (Tabla 1). Una mayor proporción de estudios RFH poseían grupo control, aunque las diferencias no alcanzaron significación estadística. El seguimiento retrospectivo fue más habitual en estudios RFH que en los estudios OP-2003/2007, mientras que en estos predominaba el diseño longitudinal prospectivo sin grupo control (p = 0,001) (Tabla 2). Respecto a otros aspectos metodológicos relevantes (justificación del tamaño muestral, el ámbito internacional de los estudios y su realización en más de un centro de investigación) los estudios RFH presentaban resultados más discretos que los encontrados para los estudios OP-2003/2007 (Figura 1), resultando la diferencia estadísticamente significativa (p < 0,05) en todas las comparaciones. No se encontraron diferencias entre ambos grupos respecto al porcentaje de estudios longitudinales con seguimiento mayor de 12 meses. Los aspectos éticos relevantes se contemplaron con mayor frecuencia en los estudios OP-2003/2007 que en los RFH (Tabla 3). El plan de trabajo del estudio se detallaba con mayor frecuencia en los protocolos de los estudios OP-2003/ 2007 que en los estudios RFH (83% vs. 55% respectivamente), así como el procedimiento de notificación de sospechas de reacciones adversas en los estudios prospectivos (93% vs 14%, respectivamente). El 60% de los estudios OP-2003/ 2007 preveía la presentación de informes de seguimiento a las autoridades sanitarias y el 79% del total, un informe final. En los estudios RFH, incumplían prácticamente en su totalidad estas obligaciones. De los 247 estudios que requerían el dictamen favorable por parte de las comunidades autónomas correspondientes al período 2003/2007, había constancia de solicitud de autorización para el 67,6%. Sin embargo, para ninguno de los RFH había constancia del cumplimiento de dicho requerimiento. Tras la revisión de los artículos publicados en la revista Farmacia Hospitalaria, se catalogaron como EPA 52 estudios (PFH). De estos, ninguno había sido registrado por la AEMPS: 20 estudios (38,5%) hubieran sido considerados EPA de seguimiento prospectivo (EPA-SP) y por tanto deberían haber sido autorizados por las autoridades sanitarias y 32 estudios (57,7%) hubieran sido clasificados como EPA con otros diseños diferentes al seguimiento prospectivo (EPA-OD). Todos se desarrollaron en ámbito nacional, 8 estudios (15,3%) fueron multicéntricos y el historial clínico fue la fuente de información de la mayoría de ellos (28 [53,8%]). En total estos estudios incluyeron 10202 pacientes (mediana = 73; p25 = 43, p75 = 242), siendo este dato ostensiblemente menor que el número de pacientes previsto incluir en estudios RFH (p = 0,0147). Los objetivos más estudiados fueron, en este orden, seguridad, investigación de resultados y utilización de medicamentos (Tabla 1). En cuanto a su diseño (Tabla 2), cabe destacar que entre los EPA longitudinales un 3,8% disponía de grupo control. Respecto a otros aspectos metodológicos (Figura 1), pocos estudios indicaban haber calculado el tamaño muestral o eran multicéntricos, siendo los resultados obtenidos más discretos a los correspondientes a estudios RFH (p < 0,05 en ambas comparaciones). El 61,5% se desarrolló en atención especializada y el 36,5% en pacientes hospitalizados. Al igual que en los estudios RFH, los grupos terapéuticos más estudiados fueron los antineoplásicos e inmunomoduladores (30,7%) y los antiinfecciosos (19,2%). Discusión Los resultados globales del estudio permiten concluir que la calidad ética y metodológica de los estudios RFH y PFH es claramente mejorable. Desde un punto de vista metodológico, la participación de un único investigador, centro o país en un estudio dificulta la representación de la diversidad de criterios clínicos y compromete su objetividad, por ello, el bajo número de estudios multicéntricos y/o internacionales podría indicar que muchos de ellos carecían de la representatividad adecuada para la generalización posterior de sus resultados10,11 . A su vez, son numerosos los estudios RFH y PFH que no presentan cálculo del tamaño muestral o poder estadístico, a pesar de ser parámetros que han de ser estimado antes del inicio de cualquier estudio que persiga el contraste estadístico de hipótesis. La precisión de los resultados puede verse comprometida en caso de no haberse previsto un tamaño muestral adecuado que posibilite alcanzar los objetivos previstos12,13. A esto último se une que un gran número de estudios no presentaban la descripción de un plan de trabajo, sin el cuál, es difícil realizar una estimación "a priori" de los recursos (humanos, sanitarios, económicos, informáticos, bibliográficos, etc..) necesarios para desarrollar el proyecto con garantías14 . Un aspecto positivo observado en los RFH es el número considerable de estudios que presentaban un grupo control, siendo mayor su proporción si lo comparamos con los estudios OP-2003/2007. El contraste de hipótesis en un estudio, hace necesaria la presencia de un grupo comparador que permita determinar la asociación y relación de causalidad entre el efecto observado y la exposición a un determinado medicamento. Dado que gran parte de los estudios RFH pretenden valorar aspectos de eficacia y seguridad de los medicamentos, parece razonable que muchos de ellos presenten grupo control para permitir su correcta evaluación15-18. Por este motivo, en los estudios RFH se considera la presencia de grupo control como un factor positivo que contribuye a una mayor calidad metodológica. En cuanto a la calidad ética de los estudios RFH, tan sólo la mitad de ellos indicaba su intención de solicitar la revisión por un CEI o acreditaban haberlo solicitado, aunque el hecho de no indicarlo en el protocolo no siempre implica que finalmente no se haya solicitado dicha revisión. Los participantes en estudios observacionales, manifiestan a menudo que no se les proporciona la suficiente información antes de la participación en un estudio y su percepción de la negativa a participar es que puede afectar a sus cuidados médicos. Por este motivo, es esencial que la hoja de información y el consentimiento informado sean legibles y comprensibles para el paciente, atendiendo a su formación y nivel sociocultural, respetando a su vez la confidencialidad de los datos19,20. El CEI es el encargado de garantizar el cumplimiento de estos aspectos, asegurando la protección de los derechos, seguridad y bienestar de los participantes en un estudio de investigación, representando por tanto el primer y principal paso para la revisión independiente de la calidad científica y ética de las propuestas de investigación, antes de su implementación5. Por todo ello, su revisión se considera esencial. Dado que un elevado número de estudios no contaban con el dictamen favorable de un CEI, se registró un elevado porcentaje de estudios RFH que no garantizaban el cumplimiento de los derechos mínimos de los participantes exigibles a este tipo de investigaciones (no se les solicitaba consentimiento para su inclusión y/o no se les informaba de la utilización de sus datos o de sus derechos de acceso, rectificación, cancelación y oposición sobre los mismos...). Aunque es significativo el número de estudios registrados en la AEMPS promovidos por los SFH, el hecho de que no se haya registrado ninguno de los PFH, sugiere un desconocimiento de la normativa que aplica a los estudios observacionales por parte de los farmacéuticos de hospital. Por otro lado, cabría pensar que la complejidad de los procedimientos administrativos y la alta carga burocrática podrían haber influido en el incumplimiento detectado9. Por otra parte, ninguno de los estudios RFH solicitó autorización a las autoridades competentes cuando por sus características les resultaba exigible. Además la remisión anecdótica de informes de seguimiento o finales a las autoridades sanitarias o no prever la notificación al Sistema Español de Farmacovigilancia de las sospechas de reacciones adversas graves detectadas en el transcurso de los estudios prospectivos, subrayan el desconocimiento de la normativa. Una de las finalidades de la Orden SAS/3470/20095, así como del resto de normativa aplicable, es mejorar la calidad científica, metodológica y consideraciones éticas de los estudios observacionales con medicamentos de uso humano. De la comparación entre los estudios RFH y PFH se puede concluir que aquellos registrados en la AEMPS presentan una calidad metodológica superior a los no registrados, en aspectos tan relevantes como presencia de grupo control, justificación del tamaño muestral y magnitud de los estudios teniendo en cuenta el número de pacientes a incluir o incluidos. Este trabajo presenta ciertas limitaciones. Una de ellas es que para determinar la calidad de los estudios promovidos por SFH pero no registrados por la AEMPS se han tomado como muestra los artículos publicados en la revista Farmacia Hospitalaria. No obstante, aunque no exista certeza de su representatividad, parece razonable pensar que los estudios publicados en este medio caracterizan suficientemente la labor investigadora de los SFH realizada al margen de la normativa. Por otra parte, los protocolos analizados podrían ser diferentes del estudio realizado cuando en el trascurso del mismo se produjeran cambios que afectaran a aspectos fundamentales del protocolo y no se comunicaran los cambios a través de enmiendas al protocolo como indica la norma. En cuanto a la comparación entre los estudios RFH y aquellos realizados por otros promotores, si bien la información de ambos grupos se puede considerar homogénea, hubiera sido deseable realizarla para períodos idénticos. No obstante, se utilizaron diferentes períodos por considerarse que en ambos la normativa aplicable presentaba requisitos similares21 . Teniendo en cuenta todo lo anterior, existen aspectos administrativos, metodológicos y éticos de los EPA promovidos por parte de los SFH que deben ser considerados según la normativa, de modo que permitan al farmacéutico de hospital aportar, de acuerdo con su formación y estatus en la práctica clínica, la información sobre el medicamento que de él espera recibir la comunidad científica y la población. A su vez, se deben incorporar los mecanismos necesarios para la adecuación de estos estudios a la normativa vigente, de modo que se garantice la protección de los derechos de los pacientes participantes. Conflicto de intereses Los autores declaran no tener ningún conflicto de intereses. Las opiniones expresadas en este trabajo son responsabilidad de los autores por lo que no reflejan necesariamente el punto de vista de los organismos en los que trabajan. Financiación Ninguna. Los autores declaran no haber publicado previamente el trabajo, ni se encuentra en proceso de revisión de otra revista. Bibliografía 1.Pardo Gracia C. Sagales Torra M, Oms Arias M, Mas Lombarte MP Evaluación de la atención farmacéutica en la prescripción de medicamentos. Farm Hosp.1995;19:133-5. [ Links ] 2.Jiménez Torres N, Cimente Martí M, Juan Colomer J, Pérez Peiró C. Metodologías para la selección de medicamentos en el hospital. Farm Hosp. 2000;24:1-11. [ Links ] 3.Moreira Lima Gamboa M, Tesainer Bruneto A, Ferreira Dos Santos ME, Gregianin L. El papel del farmacéutico en los ensayos clínicos. Farm Hosp. 2011;35:341-2. [ Links ] 4.Sacristán JA, Bolaños E. Papel de los servicios de farmacia en la realización de ensayos clínicos: punto de vista de la industria farmacéutica. Farma Hosp. 1995;19:364-7. [ Links ] 5.Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano. B.O.E. núm. 310, de 25 de diciembre de 2009, pp. 109761-109775. [ Links ] 6.Larios D. Responsabilidad ética y legal del farmacéutico hospitalario. Farm Hosp. 2011;35:287-8. [ Links ] 7.Ordovás Baines J. Investigación clínica en España y servicios de farmacia hospitalaria. Farma Hosp. 1996;20:189-91. [ Links ] 8.Pérez Martín A, Rol de la Morena MJ, Mareque Ortega M, Gómez Gómez M, Gómez Gómez C, Díaz de Cerio M. Efecto sobre el consumo de recursos hospitalarios de un programa de atención geriátrica domiciliaria en personas ancianas con patología cardiorrespiratoria muy evolucionada. Rev. Esp. Salud Publica 2001;75:559-67. [ Links ] 9.de la Fuente Honrubia C, Macías Saint-Gerons D, Vargas Castrillón E, de Abajo Iglesias FJ. Non-interventional post-authorisation studies in Spain: impact of the 2002 regulation. Med Clin (Barc). 2010 S;135:423-27. [ Links ] 10.Golder S, Loke YK, Bland M. Meta-analyses of adverse effects data derived from randomised controlled trials as compared to observational studies: methodological overview. PLoS Med. 2011;8(5):e1001026. [ Links ] 11.Blumenstein BA, James KE, LInd BK, Mitchell HE. Functions and organization of coordinating centers for multicenter studies. Controlled Clinical Trials. 1995;16:4S29S. [ Links ] 12.Marrugat J, Vila J, Pavesis M, Sanz F. Estimación del tamaño de la muestra en la investigación clínica y epidemiológica. Med Clin (Barc). 1998;11 1:267-76. [ Links ] 13.Sabaté D. El tamaño de muestra en los estudios de eficacia. Rev Epidem. Med. Prev. 2003;1:27-30. [ Links ] 14.Hahn M, Ruppert T, Bethke TD, Hundt F. Results of a survey on applied quality standards in non-interventional studies among the members of the German Association of Research-based Pharmaceutical Companies. Ger Med Sci. 2010;8:Doc 29. [ Links ] 15.Vandenbroucke JP, Von Elm E, Altman DG, Gotzsche PC, Mulrow CD, Pocock SJ, et al. Mejorar la comunicación de estudios observacionales en epidemiología (STROBE): explicación y elaboración. Gac Sanit. 2009;23:158. [ Links ] 16.Freedland KE, Mohr DC, Davidson KW, Schwartz JE. Usual and unusual care: existing practice control groups in randomized controlled trials of behabioral interventions. Psychosom Med. 2011;73:323-35. [ Links ] 17.Hochman M, McCormick D. Characteristics of published comparative effectiveness studies of medications. JAMA. 2010;303(10):951-8. [ Links ] 18.Concato J, Lawler EV, Lew RA, Gaziano JM, Aslan M, Huang GD. Observational methods in comparative effectiveness research. Am J Med. 2010;123 Suppl 1:16-23. [ Links ] 19.Kiguba R, Kutyabami P, Kiwuwa S, Katabira E, Sewankambo NK. Assessing the quality of informed consent in a resource-limited setting: A cross-sectional study. BMC Med Ethics. 2012;13:21. [ Links ] 20.Vreeman R, Kamaara E, Kamanda A, Ayuku D, Nyandiko W, Atwoli L, et al. A qualitative study using traditional community assemblies to investigate community perspectives on informed consent and research participation in western Kenya. BMC Med Ethics. 2012;13:23. [ Links ] 21.Circular 15/2002 de 10 de Octubre. Procedimientos de comunicación en materia de Farmacovigilancia de medicamentos de uso humano entre la Industria Farmacéutica y el Sistema Español de Farmacovigilancia de medicamentos de uso humano. Agencia Española del Medicamento. Disponible en: http://www.aemps.gob.es/informa/circulares/medicamentosUsoHumano/seguridad/2002/home.htm [ Links ] Recibido: 27/05/2013.
2Unidad de Ensayos Clínicos de Fase I de Oncología. Fundación Jiménez Díaz. Madrid.
3Servicio de Farmacia de la Dirección Asistencial Centro. Servicio Madrileño de Salud.
Métodos: Se identificaron los estudios promovidos por los SFH registrados en la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) durante 2009-2011 y/o publicados en la revista Farmacia Hospitalaria en el mismo período. Se analizaron los aspectos éticos y metodológicos más relevantes. Con el fin de conocer las diferencias de los estudios promovidos por los SFH respecto a estudios realizados por otros promotores, se compararon con los estudios registrados durante 2003-2007.
Resultados: Se registraron en la AEMPS 22 estudios promovidos por SFH, que comparados con aquellos realizados por promotores diferentes, mostraron un menor cumplimiento de los aspectos éticos recogidos en la normativa, así como resultados más discretos y estadísticamente significativos (p < 0,05), respecto a justificación del tamaño muestral (41,5% vs 80%) o ámbito internacional (0% vs 24%). Respecto a los estudios publicados en la revista Farmacia Hospitalaria (n = 52), ninguno fue registrado en la AEMPS. En comparación con los estudios registrados promovidos por SFH, presentaron menor calidad metodológica, en aspectos tales como presencia de grupo control (3,8% vs 27,3%) (p = 0,0072) o justificación del tamaño muestral (19,2% vs 42,8%) (p < 0,05).
Conclusión: Existen aspectos administrativos, metodológicos y éticos de los estudios promovidos por los SFH que deben ser mejorados según la normativa. El registro en la AEMPS, parece tener un efecto positivo en el rigor científico y ético de los protocolos de investigación.
Methods: HPD promoted studies in the 2009-2011 period included in the Spanish Agency of Medicines and Medical Devices (AEMPS) registry and/or published in "Farmacia Hospitalaria" were identified. The most relevant ethical and methodological characteristics were analyzed. Studies promoted by HPD were also compared with studies not promoted by HPD.
Results: Twenty two studies promoted by HPD, and registered in the AEMPS were identified. Within the registered studies HPD promoted studies had lower sample size estimation (41,5% vs 80%) and international scope (0% vs 24%) compared to non HPD promoted studies with significant differences (p < 0,05). None of the published studies in the journal Farmacia Hospitalaria have been registered in the AEMPS and had lower methodological quality than the registered studies promoted by HPD in characteristics such as presence of control group (3,8% vs 27,3%) (p = 0,0072) and the sample size estimation of (19,2% vs 42,8%) (p < 0,05).
Conclusion: The management and the methodological and ethical characteristics of the studies promoted by HPD should be improved according to the regulation. The registration in the AEMPS might have a positive impact on the quality of these research protocols.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Diana González Bermejo
E-mail: dgonzalezb@aemps.es
Aceptado: 29/09/2013.

