










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedifam
versión impresa ISSN 1131-5768
Medifam vol.11 no.8 ago./sep. 2001
NOTA CLÍNICA
Un síndrome de Patau con una supervivencia que supera los pronósticos
L. Sierra Santos, C. Álvarez Herrero, L. Gil Sánchez*, E. Sierra Santos**
*Médico de Familia. *Residente de Familia.**Diplomada Universitaria en Enfermería.
Centro de Salud de Manzanares el Real y V Centenario. Área 5. Madrid
RESUMEN
La trisomía del cromosoma 13 o el Síndrome de Patau es una aneuploidía infrecuente presente con una incidencia de uno entre cada 20.000 nacidos vivos y que sólo en rarísimas ocasiones permite la supervivencia de estos pacientes más de un año.
Presentamos el caso de una paciente con un mosaicismo celular del 93% de sus células trisómicas y el 7% euploides que ha alcanzado la edad adulta a pesar de sus severas malformaciones.
Palabras clave: Trisomía 13. Malformaciones congénitas. Síndrome de Patau. Aneuploidía.
Patau's syndrome, a survival that gets over the prognosis
ABSTRACT
The trisomy 13 or Patau´s Syndrome is an infrecuent aneuploidy which has an incidence of one among 20,000 liveborn, that only in rare circumstances lets these patients live more than a year.
We present the case of a patient with mosaicism of 93% trisomic cells and the 7% euploid ones who has reached the adulthood in spite of her serious somatic malformations.
Key words: Trisomy 13. Congenital malformation. Patau´s Syndrome. Aneuploidy.
INTRODUCCIÓN
La trisomía del cromosoma 13 o Síndrome de Patau es una cromosomopatía que ocurre en uno de cada 20.000 nacidos vivos, aunque es más frecuente en abortos espontáneos y mortinatos1. Se asocia a graves malformaciones congénitas de la línea media corporal, anomalías oculares, en las extremidades y alteraciones viscerales como malformaciones cardiacas, renales y cerebrales. Estas malformaciones marcan el pronóstico que generalmente es infausto, con una supervivencia rara vez superior al año de vida2.
Presentamos el caso de una paciente con una trisomía 13 que ha alcanzado la edad adulta a pesar de sus graves malformaciones.
OBSERVANCIA CLÍNICA
Se trata de una paciente nacida en enero de 1975 de padres no consanguíneos y sanos. Madre de 29 años de edad, secundípara y secundigesta. Hermano sano. Como único antecedente la madre refiere que presentó cefaleas intensas durante el embarazo y tomó una medicación que no sabe precisar. Al quinto o sexto mes de gestación notó movimientos fetales de poca intensidad y el parto fue a término y eutócico. La niña pesó al nacer 2,950 g y midió 48 cm. Presentó dificultad respiratoria a los pocos minutos del nacimiento, por lo que fue ingresada en la unidad de reanimación neonatal y dada de alta a los pocos días. Los padres refieren que presentaba una succión débil durante las primeras semanas de lactancia y movimientos laterales de cabeza persistentes y repetitivos, por lo que consultaron a su pediatra, que envió a la paciente al hospital para realizar un estudio genético por detectar múltiples malformaciones. Al mes de vida pesaba 3,800 g (percentil 25-50) y medía 50 cm (percentil 10). Presentaba una facies con oblicuidad antimongólica, nariz dorso convexa y ancha, orejas de implantación baja, pobre desarrollo del pabellón auricular y el paladar ojival (Fig. 1). Tenía hexadactilia cicatricial bilateral en el borde cubital de las manos. Los dedos de la paciente presentaban una flexión permanente (camptodactilia) y una curvatura lateral de los mismos (clinodactilia) (Fig. 2). En el pie izquierdo presentaba sindactilia entre el segundo y el tercer dedo, un angioma en el dorso, así como un calcáneo prominente. El primer dedo de los pies era grande y de forma cúbica (Fig. 3). El desarrollo neurológico a esa edad era aparentemente normal, así como las analíticas de sangre, orina y del líquido cefalorraquídeo. En el neumoencefalograma se visualizó una alteración morfológica de los ventrículos laterales con forma cornada en las porciones superoexternas y una ligera hipogenesia del cuerpo calloso. El trazado del electroencefalograma era normal así como la exploración urológica y cardiaca. En la radiografía de tórax se apreció una osificación anormal del esternón. En la exploración oftalmológica se descubrió un coloboma bilateral del iris con restos de masa cristalina en reabsorción. No presentaba fijación de la mirada. El cariotipo realizado dio como resultado una trisomía del grupo D (13-15) primaria. El cariotipo de los padres era normal. Con el diagnóstico de Síndrome de Patau con posibles crisis comiciales la paciente comenzó tratamiento con fenitoína.

Figura 1. Cara de la paciente con nariz ancha, pabellones auditivos de implantación baja, cuello con abscesos y trayectos fistulosos. Se aprecia la gran hipertrofia gingival.

Figura 2. Mano de la paciente con campto y clinodactilia. Hexadactilia cicatricial en el borde cubital.
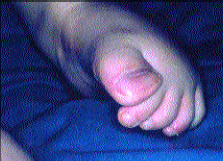
Figura 3. Pie de la paciente con el primer dedo de forma cúbica, resto de los dedos superpuestos.
Al año de vida se mantenía sentada, a los dos años decía monosílabos, parecía ver luz y oír poco, reconocía la voz de los padres aunque padecía una encefalopatía grave. En la radiografía de muñeca la edad ósea correspondía a una niña de cuatro meses. La dentición era completa con defectos en el esmalte achacables a la medicación con fenitoína. Se realizó una tomografía axial computerizada de las órbitas donde existía diferencia de densidad de los globos oculares que podía corresponder a la primera fase de un retinoblastoma. Se decidió una actitud expectante ante este hecho que nunca llegó a confirmarse. Tras cuatro meses, una nueva exploración ocular permitió ver que la imagen del escáner correspondía a la persistencia de la arteria hialoidea y a la formación de una catarata posterior. Desde los siete años el tratamiento de las crisis comienza a ser el fenobarbital. A los diez años daba algunos pasos con dificultad, parecía ver poco y no hablaba. No comprendía ni sintonizaba con el medio aunque emitía ruidos aislados, sin relación con estímulos externos. Presentaba incontinencia de esfínteres y el retraso mental era muy profundo.
Desde los seis años de edad la paciente comenzó a padecer abscesos de repetición en las axilas e ingles con fístulas que han necesitado antibioterapia repetitiva. A los once años precisó un ingreso por este motivo y fue estudiada en el Servicio de Inmunología donde se le diagnosticó una alteración en la movilidad y quimiotaxis de los neutrófilos típica de este síndrome. Comenzó tratamiento con vitamina C. Los abscesos han sido desde ese momento foco importante de fiebre y se ha necesitado la participación de los Servicios de Dermatología y Cirugía Plástica para el desbridamiento de los trayectos fistulosos. Así mismo, se han realizado cultivos de los abscesos y antibiogramas en los episodios febriles, cultivándose numerosas bacterias aerobias, anaerobias y hongos. Se ha constatado la presencia de anemia ferropénica que se ha imputado también al abundante sangrado crónico de las colecciones purulentas.
A los dieciocho años de edad la paciente ingresó de nuevo en el hospital por una sepsis de origen urinario, donde se diagnosticó además una insuficiencia renal leve y una hepatopatía crónica avanzada con citolisis y colestasis. Presentaba también desnutrición. En la analítica destacaba: hemoglobina 9,2 g/dl; VCM 75,7 fL; 14.500 leucocitos con predominio de neutrófilos; GOT 444 U/l; GPT 347 U/l; GGT 334 U/l; fosfatasa alcalina 535 U/l; LDH 733 U/l; bilirrubina total 14,7 mg/dl y conjuada 11,3 mg/dl; albúmina 2,9 g/dl y proteínas totales 6,2 g/dl. La ecografía abdominal permitió visualizar una vesícula acodada con barro y entre la misma y la porta se detectó un asa intestinal malformada e interpuesta a ese nivel. En el hígado aparecieron múltiples granulomas calcificados. Se apreció también un bazo supernumerario de 1 cm en el polo inferior esplénico.
A los veinte años de edad, a pesar del tratamiento, presentó un estatus epiléptico por lo que éste fue modificado y se introdujo de nuevo la fenitoína. Pasados tres años la paciente presentaba una hipertrofia gingival enorme que le impedía comer con normalidad (Fig. 1) y que precisó la valoración del Equipo de Maxilofacial y la sustitución del fármaco causante. Actualmente está en tratamiento con carbamacepina y fenobarbital. Presentó además un hipotiroidismo subclínico no susceptible de tratamiento aún. A los ventiún años de edad presentó la menarquia aunque sus reglas han sido irregulares desde ese momento debido a la anemia y a su nivel de desnutrición.
El cariotipo de la paciente fue repetido en el año 1998 y se perfiló con garantías suficientes (tras revisar más de cien leucocitos) que se trataba de un cariotipo femenino con mosaicismo del 7% de células normales y una trisomía del cromosoma 13 en el 93% de las mismas (Fig. 4).

En la actualidad la paciente tiene 26 años y sobrevive aún con sus múltiples complicaciones, un retraso mental profundísimo y el deterioro de las escasas facultades que tuvo. Ya no camina y precisa de cuidados constantes domiciliarios.
DISCUSIÓN
Desde que Tjio y Levan descubrieron en 1956 el número de cromosomas en la especie humana y tres años después fuera descrita por Lejeune la primera trisomía (el Síndrome de Down), se han desarrollado numerosas técnicas citogenéticas para identificar con detalle los cromosomas y sus segmentos, gracias a los cuales se obtienen diagnósticos clínicos más precisos y consejos genéticos más exactos2.
La trisomía 13 fue descrita por Patau en 19603 a la vez que Edward describió la trisomía 184. Ninguno de los dos autores pudo en aquél momento identificar el cromosoma exacto responsable de dichas aneuploidías debido a los defectos de la técnica empleada, pero sí establecieron con detalle las malformaciones características de dichos síndromes y la escasa supervivencia asociada a ellos, que no ha mejorado a pesar de los avances de la Ciencia.
El Síndrome de Patau tiene una incidencia de1 entre 20.000 nacidos vivos, aunque en abortos espontáneos es 100 veces más frecuente2. Es ligeramente más frecuente en mujeres, al igual que la trisomía 18. No así la trisomía 21, lo que hace pensar que se produce por mecanismos distintos o por una selección a favor de un sexo por motivos aún desconocidos5. Se trata de una aneuploidía producida por la no-disyunción de dos cromátidas hermanas durante la primera división meiótica que genera alguna célula con un cromosoma 13 más que surge de forma espontánea o por influencias ambientales3,6. Como todas las trisomías se ha relacionado la edad materna avanzada como un factor predisponente. El 63% de las trisomías ocurren en madres de más de 35 años7.
Las malformaciones1,3,8,9 (Tabla I) se producen por una sobreexpresión o disbalance del cromosoma 131,10. Algunos autores consideran, además, que pueden existir agentes exógenos, desconocidos aún, que influyan en la expresión de las malformaciones1.

Los pacientes con Síndrome de Patau tienen una supervivencia muy escasa. Aproximadamente el 28% muere en la primera semana de vida; el 44% en el primer mes y el 86% en el primer año. Sólo el 5% sobrevive más de 3 años11. La supervivencia media es de 2,5 días para Goldstein y de 90 días para Taylor, aunque hay casos aislados que llegan a la adolescencia12 y excepcionalmente a la edad adulta11,13.
El pronóstico de vida se relaciona claramente con la gravedad de las malformaciones cerebrales, renales y cardiacas.
La supervivencia en el Síndrome de Patau es más larga en individuos mosaicos, esto es, en pacientes que tienen líneas celulares trisómicas y líneas celulares normales1,10, como es el caso de nuestra paciente. Sin embargo, el mosaicismo en el Síndrome de Patau es una rareza y parece existir sólo en el 5% de los casos14. Petit en 1994 refirió un mosaico con una supervivencia de 38 años, pero el porcentaje de las células trisómicas era únicamente del 1%13. Llama poderosamente la atención que nuestra paciente presente una supervivencia tan larga con un porcentaje de células trisómicas del 93% ya que el fenotipo encontrado habitualmente depende de la fracción de células anormales y de la distribución en los tejidos. En el caso presentado desconocemos la distribución de la trisomía en otros tejidos, ya que únicamente se ha realizado el cariotipo en los tejidos sanguíneos. En mosaicos con trisomía 13 no es frecuente, sin embargo, que haya rasgos asociados a la trisomía 13 pura y pocos casos presentan polidactilia u holoprosencefalia. Asumimos, por tanto, que en el caso presentado las líneas celulares afectadas son muy extensas, ya que el fenotipo es muy próximo a una trisomía pura. Es raro que en cualquier mosaicismo del cromosoma 13 existente la inteligencia esté intacta, aunque el porcentaje de linfocitos afectados por la trisomía no puede ser predictor de la misma14.
Recientemente se ha acuñado el término de pseudotrisomía 13, que plantea el diagnóstico diferencial con el Síndrome de Patau. Se trata de individuos con malformaciones similares (holoprosoencefalea y polidactilia) pero con cariotipo normal. Es más frecuente en la raza judía y en padres consanguíneos, lo que hace pensar en una herencia autosómica recesiva15.
En las series de pacientes recientes la supervivencia es menor que la publicada hace 30 años, debido probablemente a que en la actualidad el diagnóstico precoz de las malformaciones fetales aumentan la frecuencia de las interrupciones voluntarias del embarazo. Existe, además, un registro mayor de los casos con supervivencia baja y la actitud de los profesionales sanitarios ya no tiende a intervenciones quirúrgicas ni a medidas vitales extraordinarias con estos pacientes, de los que bien se conoce su infausto pronóstico12.
Presentamos este caso por el interés que entre la Comunidad Científica despierta y porque desde hace años sus principales cuidadores son sus familiares y los profesionales del Equipo de Atención Primaria.
AGRADECIMIENTOS
Estamos agradecidas a la Dra. Isidora López Pajares por la laboriosa realización del cariotipo y a D. José Segovia Bollo, nuestro desinteresado fotógrafo.
CORRESPONDENCIA:
Lucía Sierra Santos
C/ Maestro Barbieri, 11 bajo D
28100 Alcobendas
Madrid
Bibliografía
1. Zoll B, Wolf J, Lensing-Hebben D, Pruggmayer M, Torpe B. Trisomy 13 with an 11-years survival. Clin Genet 1993; 43 (1): 46-50. [ Links ]
2. Hirschhorn K. Enfermedades prenatales. En: Berhrman RE. Nelson. Tratado de Pedriatía. 14ª ed. Madrid: WB Saunders Company, 1992; 319-66. [ Links ]
3. Patau K, Smith DW, Therman E, Inhorn SL, Wagner HP. Multiple congenital anomaly caused by an extra autosome. Lancet 1960; 1: 790-3. [ Links ]
4. Edwards JH, Harnden DG, Cameron AH, Crosse VN, Wolff OH. A new trisomic syndrome. Lancet 1960; 1: 787-9. [ Links ]
5. Huether CA, Martin RL, Stoppelman SM, D´souza S, Bishop JK, Torfs CP, et al. Sex ratios in fetuses and liveborn infants with autosomal aneuploidy. Am J Med Genet 1996; 63 (3): 492-500. [ Links ]
6. German J. Aspectos citogenéticos de la enfermedad humana. En: Fauci AS, Braunwald E, Isselbacher KJ, Wilson JD, Martin JB, Kasper DL, et al. Harrison. Principios de Medicina Interna. 14ª ed. Madrid: McGraw-Hill-Interamericana, 1998; 450-8. [ Links ]
7. Salihu HM, Boos R, Schmidt W. Antenatally detectable markers for the diagnosis of autosomally trisomic fetuses in at risk pregnancies. Am J Perinatol 1997; 14 (5): 257-61. [ Links ]
8. Ashraf Aziz M. Muscular anomalies caused by delayed development in human aneuploidy. Clin Genet 1981; 19: 111-6. [ Links ]
9. Chen CP, Liu FF, Jan SW, Su TH, Lan CC. A concealed penis mimicking penile agenesis in an infant with trisomy 13. Clin Genet 1996; 50: 159-8. [ Links ]
10. Hirschhorn K, Cooper HL. Chromosomal aberrations in human disease. Am J of Med 1961; 31: 442-68. [ Links ]
11. Singh. Trisomy 13 (Patau´s syndrome): a rare case of survival into adulthood. J Ment Defic Res 1990; 34: 91-3. [ Links ]
12. Goldstein H, Nielsen KG. Rates and survival of individuals with trisomy 13 and 18. Clin Genet 1988; 34: 366-72. [ Links ]
13. Petit P, Fryns JP. Normal/trisomy 13 mosaicism in a 38 year-old male. Genetic Couns 1994; 5: 311-4. [ Links ]
14. Delatycki M, Gardner RJ. Three cases of trisomy 13 mosaicism and a review of the literature. Clin Genet 1997; 51 (6): 403-7. [ Links ]
15. Cohen MM, Gorlin RJ. Pseudo-trisomy 13 syndrome. Am J Med Genet 1991; 39 (3): 332-5. [ Links ] [Abstract].

