










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedifam
versión impresa ISSN 1131-5768
Medifam vol.13 no.1 ene. 2003
NOTA CLÍNICA
Un caso de lesiones ampollosas generalizadas
M. Repiso Moreno, R. Daroca Pérez1, Mª. J. Elizondo Pernaut2
Especialista en Medicina Familiar y Comunitaria. Servicio de Medicina Interna.
1Servicio de Medicina Interna. Complejo Hospitalario San Millán- San Pedro. Logroño
2Diplomada en Enfermería. Hospital de Navarra. Pamplona.
| RESUMEN El penfigoide ampolloso es una enfermedad cutánea ampollosa que predomina en personas de edad avanzada. Su curso es crónico, caracterizado por periodos de exacerbación y remisión. Las lesiones son típicamente ampollas localizadas en tronco y superficies flexoras de extremidades y, en ocasiones afectan también a mucosas. El diagnóstico está basado en la combinación de hallazgos clínicos, histológicos e inmunopatológicos. El tratamiento clásico son los corticoides tópicos o sistémicos. Se presenta el caso de un varón de 50 años con una forma extensa de penfigoide ampolloso. Palabras clave: Penfigoide ampolloso. Enfermedad ampollosa. Glucocorticoides. | A case of extensive bullous pemphigoids ABSTRACT The bullous pemphigoid is an autoinmune blistering skin disease predominantly seen in the elderly persons. It is a cronic disease characterised by periods of exacerbation and remission. Usually lesions are blisters localized at trunk and the flexor surfaces of the extremities and sometimes involves oral mucosa. The diagnosis is based on the combination of clinical, histologic and inmunopathologic findings. The standard treatment is topical o systemic glucocorticoids. We report a case of a 50 years old man with an extensive bullous pemphigoid. Key words: Bullous pemphigoid. Blistering disease. Glucocorticoids. |
Recepción: 19-04-02
Aceptación: 19-09-02
INTRODUCCIÓN
La aparición de lesiones ampollosas es un hecho relativamente frecuente que incluye desde patologías banales autolimitadas hasta procesos graves con elevada morbimortalidad. En algunos casos la ampolla es un signo añadido, pero existe un grupo de enfermedades entre las que figura el penfigoide ampolloso, en las cuales esta lesión constituye el verdadero sustrato patológico.
CASO CLÍNICO
Varón de 50 años sin antecedentes patológicos de interés. Fumador de 40 cigarrillos/día y bebedor moderado.
Acude por presentar desde hace 10 días placas eritematoedematosas y lesiones vesiculoampollosas tensas de contenido serohemático sobre base eritematosa, pruriginosas, localizadas a nivel de tronco y extremidades, respetando palmas, plantas, cara y cuero cabelludo. El inicio fue a nivel de región proximal de extremidades, extendiéndose posteriormente. En todo momento permanece afebril.
No refiere toma de fármacos ni sintomatología digestiva.
En la exploración física la auscultación cardiopulmonar era normal, al igual que el abdomen, presentando a nivel cutáneo las lesiones antes descritas, con signo de Nikolsky negativo. No se apreciaban lesiones en cavidad oral ni en otras mucosas.
En las exploraciones complementarias realizadas destacaba en la analítica una leucocitosis de 17.400 con eosinofilia (13,6%), siendo el resto del hemograma y la bioquímica normales. La radiología de tórax no mostraba hallazgos patológicos.
El paciente es remitido al Servicio de Medicina Interna para completar estudio e iniciar tratamiento.
Los anticuerpos antiendomisio fueron negativos. Los anticuerpos antimembrana basal epidérmica (por inmnufluorescencia indirecta) presentaron una positividad moderada con dilución 1/20 de tipo IgM e IgG y negatividad para IgA.
Se realizó una biopsia cutánea en la que se apreciaba una ampolla subepidérmica con infiltrado inflamatorio leve en dermis constituído por eosinófilos. La inmunofluorescencia directa (IFD) demostró presencia de depósitos de IgG en la membrana basal epidérmica.
Con el diagnóstico de penfigoide ampolloso se inició tratamiento con prednisona hasta alcanzar 100 mg/día sin evidenciar mejoría, por lo que se añadió azatioprina a dosis de 100 mg/día reduciendo progresivamente la cantidad de corticoides con buena respuesta clínica y mejoría progresiva de las lesiones. A los 6 meses presenta un buen control de la enfermedad manteniendo dosis de 100 mg/día de azatioprina y 10 mg/día de prednisona, siguiendo revisiones periódicas por parte de Atención Primaria y del Servicio de Medicina Interna.
DISCUSIÓN
El penfigoide ampolloso es una enfermedad de patogenia autoinmune caracterizada por la aparición de lesiones ampollosas subepidérmicas.
Es la enfermedad ampollosa autoinmune más frecuente en los países occidentales, con una incidencia anual de 7 casos por millón de habitantes y suele aparecer en personas mayores de 60 años, sin predominio de sexo.
En estos pacientes se da la presencia de autoanticuerpos contra el antígeno del penfigoide ampolloso, que es un componente de la membrana basal epidérmica. Dichos anticuerpos son capaces de activar la vía del complemento que es un elemento esencial en la fisiopatología del penfigoide ampolloso1. La caracterización de este antígeno ha permitido descubrir que está formado por dos polipéptidos de 230 y 180 kilodaltons.
Se han relacionado las moléculas de clase II del sistema mayor de histocompatibilidad DQB1 con una mayor susceptibilidad al penfigoide ampolloso2.
El inicio de las lesiones es polimorfo, pudiendo aparecer máculas o placas urticariformes sobre las que posteriormente se asientan ampollas de 1-3 cm de diámetro, de contenido claro o sanguinolento, tensas y pruriginosas (Fig. 1). Las localizaciones más típicas son parte baja del abdomen, axilas, ingles y superficies flexoras de las extremidades (Fig. 2). En aproximadamente la tercera parte de los casos se afecta la mucosa oral y pueden afectarse otras mucosas como la esofágica, produciendo cuadros de esofagitis. Las ampollas curan sin dejar lesiones en la piel3.
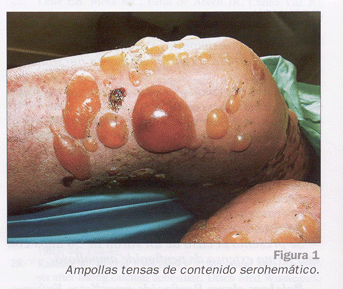

El cuadro suele ser crónico, pudiendo persistir durante meses o años y cursa con exacerbaciones y remisiones.
El signo de Nikolsky es negativo.
El diagnóstico se basa en la biopsia cutánea en la que aparecen ampollas subepidérmicas con un infiltrado inflamatorio dérmico en el que predominan los eosinófilos. La epidermis se encuentra indemne. El estudio inmunopatológico por IFD demuestra depósitos de IgG y/o C3 en la membrana basal epidérmica.
Por inmunofluorescencia indirecta se pueden detectar en sangre anticuerpos antimembrana basal epidérmica de tipo IgG en aproximadamente el 70% de los casos4. Los niveles de autoanticuerpos no tienen relación con la actividad de la enfermedad, aunque hay autores que relacionan un nivel elevado de anticuerpos frente al antígeno de 180 kilodaltons con formas más severas de la enfermedad.
El diagnóstico diferencial hay que hacerlo con enfermedades como el pénfigo, dermatitis herpetiforme, penfigoide cicatricial, herpes gestacional, epidermolisis ampollosa, porfiria cutánea tarda, eritema multiforme, dermatosis medicamentosas5 (Tabla I).

El tratamiento de elección son los corticoides sistémicos utilizando la prednisona a dosis de 0,5-1 mg/kg/día. En los casos rebeldes o con el objetivo de reducir la dosis de corticoides se pueden asociar inmunosupresores como la azatioprina o ciclofosfamida6,7.
Otros tratamientos utilizados son dapsona, micofenolato mofetil, o en casos muy rebeldes la plasmaféresis8.
En aquellos pacientes con enfermedad leve o poco extensa se pueden utilizar corticoides tópicos, aunque hay estudios recientes que afirman que los corticoides administrados de forma tópica son tan efectivos como los orales en formas moderadas y severas con menores efectos secundarios9. Esto es un hecho importante ya que esta enfermedad aparece fundamentalmente en pacientes ancianos en los que los corticoides sistémicos pueden descompensar otras patologías concomitantes.
Hay estudios que sugieren una mayor incidencia de tumores en pacientes con penfigoide ampolloso, aunque esto es un hecho controvertido10.
La mortalidad es relativamente baja, siendo del 19% en el primer año, fundamentalmente en ancianos y personas debilitadas.
CORRESPONDENCIA:
Manuel Repiso Moreno
Avda. Eulza, 77, 1º D
31010 Barañaín. Navarra
Bibliografía
1. Salmon- Ehr, Bernard P. Physiopathologie des dermatosses bulleuses auto- inmunes de la jonction dermo- épidermique. Ann Dermatol Venereol 1998; 125: 817-23. [ Links ]
2. Mascaró JM. Autoinmune blistering diseases of the skin: diseases of cell adhesion. Inmunología 2001; 20: 105-13. [ Links ]
3. Aliaga A, Alegre V. Enfermedades de la boca. En: Farreras-Rozman. Medicina Interna. 13ª ed. Barcelona: Mosby-Doyma libros, 1995. p. 46. [ Links ]
4. Sánchez-Aguilar A, Flórez A, Ginarte M. Pénfigos y penfigoides. Medicine 1999; 7: 6381-6. [ Links ]
5. Arenas R. En: Dermatología: Atlas diagnóstico y tratamiento. 1ª ed. México: Mc Graw Hill, 1987; 179-81. [ Links ]
6. Yancey K, Egan C. Pemphigoid: Clinical, Histologic, Inmunopathologic, and Therapeutic Considerations. JAMA 2000; 284: 350-6. [ Links ]
7. Yancey K, Lawley T. Enfermedades cutáneas de mecanismo inmunológico. En: Braunwald E, Fauci A, Kasper D, Hauser S, Longo D, Jameson J. Harrison. Principios de Medicina Interna. 15ª ed. Madrid: Mc Graw- Hill, 2002. p. 391-6. [ Links ]
8. Nousari H, Anhalt G. Pemphigus and bullous pemphigoid. Lancet 1999; 354: 667-72. [ Links ]
9. Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med 2002; 346: 321-7. [ Links ]
10. Lindelof B, Islam N, Ellward G, Alfors L. Pemphigoid and cancer. Arch Dermatol 1990; 126: 66-8. [ Links ]

