










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Salud Pública
versión On-line ISSN 2173-9110versión impresa ISSN 1135-5727
Rev. Esp. Salud Publica vol.73 no.5 Madrid sep. 1999
EPIDEMIOLOGIA MOLECULAR DEL VIRUS DEL SARAMPION
Rafael Fernández-Muñoz, Juan Carabaña, Montserrat Caballero, Paloma B. Liton, Beatriz M Duque, Dolores García-Villalón y María Luisa Celma.
Virología, Hospital "Ramón y Cajal"
Correspondencia
Virología, Hospital "Ramón y Cajal"
Carretera de Colmenar, Km 9
28034 Madrid.
INTRODUCCIÓN
El virus del sarampión (VS) pertenece al género de los Morbillivirus, que agrupa a los virus del moquillo canino, virus de la peste bovina, virus de los pequeños rumiantes, morbillivirus de los fócidos, y al morbillivirus de los delfines. Este género pertenece a la familia de los Paramixoviridae, que son virus RNA de una sola cadena de polaridad negativa (complementaria a los RNA mensajeros) que forma una nucleocápside helicoidal recubierta de membrana de origen celular1.
El genoma de los virus salvajes del sarampión es una molécula de RNA de 15.984 nucleótidos2, codifica por las proteínas estructurales: N (nucleocápsida), P (fosfoproteína), L (polimerasa), M (matriz), H (hemaglutinina) y F (de fusión) que se incorporan a las partículas víricas, y otras no estructurales V y C que se encuentran solamente en las células infectadas. En la figura 1 se esquematizan la estructura de virus y sus proteínas.
Figura 1.
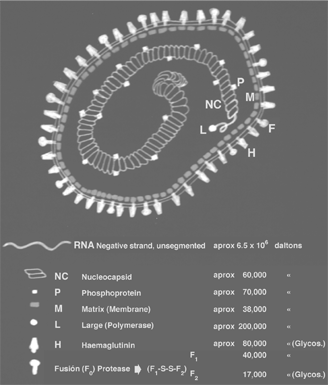
El virus de sarampión es un virus de transmisión respiratoria que infecta a humanos y primates, y del que no se han descrito serotipos.
Produce un cuadro exantemático, que va acompañado de una inmunosupresión transitoria que probablemente contribuye a una parte de las complicaciones por infecciones oportunistas. El sarampión y sus complicaciones, que pueden ser severas, actualmente está matando más de un millón de niños al año, a nivel mundial.
Al margen de las complicaciones de la infección aguda, principalmente respiratorias y neurológicas, el VS causa encefalitis de curso subagudo, que puede iniciar sus manifestaciones clínicas al cabo de meses, como la encefalitis en inmunodeficientes (MIBE), o de años o décadas, como la Panencefalitis Esclerosante Subaguda (SSPE), ambas de curso prolongado y mortal.
Se dispone de una vacuna viva atenuada que es eficaz, aunque en niños de menos de un año en países de bajo nivel sanitario no sea suficientemente efectiva, debido a anticuerpos neutralizantes maternos u otras causas asociadas no identificadas.
En nuestro laboratorio hemos procedido a determinar la secuencia de genes de virus VS recién aislados por nosotros, para excluir cualquier variación en su genoma introducida durante su cultivo. El objetivo es doble, por un lado conocer la estructura completa del genoma de virus del sarampión salvaje, ya que, hasta ahora, sólo se ha reportado el genoma de un virus atenuado vacunal, y por otro lado, definir los posibles genotipos circulantes de VS y estudiar su distribución geográfica y evolución.
En colaboración con laboratorios de Virología de Queen´s University de Belfast, Colin Dales Laboratories de Londres, Universität Würzburg, y del CDC en Atlanta, hemos definido la mayoría de los genotipos circulantes en las últimas décadas, a nivel global. En el árbol filogenético, elaborado a partir de la secuencia de la zona variable del gen N mediante un programa CLUSTAL-53, se observa que los aislados estudiados se agrupan en un número limitado de grupos, en general bien diferenciados, para los que propusimos la denominación de genotipos A,.....F. Aunque los virus de un genotipo se han aislado en distintas áreas geográficas, observamos cierto endemismo y alguno de ellos, como el F, con aislados circulantes en Madrid, parecen haberse extinguido.
Cuando elaboramos un árbol filogenético a partir de las secuencias del gen H4, se obtuvo una distribución similar para todos los aislados estudiados, excepto uno más mutado en estos genes Este resultado apoya la definición hecha de los genotipos según el gen N y, por otro lado, no indica recombinación entre los distintos genotipos de VS.
Al seguir la evolución a lo largo de las tres últimas décadas de los genotipos circulantes en un área geográfica, Madrid, hemos observado que hay sustituciones sucesivas en el genotipo que circula (figura 2). El último cambio de genotipo detectado tuvo lugar en el otoño de 1994, cuando el genotipo C2 dió paso al genotipo D3, probablemente, a partir de un caso importado de otro área todavía no identificada, como hemos mostrado evidencia que ha ocurrido para una importación y posterior difusión del genotipo C2 desde Europa a USA5.
Figura 2.
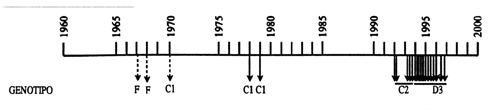
Durante los tres años en los que el VS D3 viene circulando en Madrid, hemos estudiado su evolución investigando la secuencia completa del gen H. En la serie temporal de nuestros aislados, hemos computado las mutaciones silentes y las que producen cambios de aminoácidos en la proteína (3). Con estos datos hemos podido realizar, por vez primera, una estimación de la velocidad de mutación de un gen de VS en una epidemia. Obtenemos, para el gen H un valor de 5x10 -4, cambios por año para una posición de nucleótido, que es un valor 10 a 20 veces menor de la deriva del virus de la gripe. Así, VS resulta ser un virus relativamente estable, aunque se observan virus altamente mutados como IFF95 que, aparentemente, no se fijan en la población.
Actualmente, estamos determinando en los aislados D3 la relación de mutaciones silentes o sinónimas, que no producen cambios de aminoácido, a mutaciones no sinónimas que sí producen cambios de aminoácido. El objetivo es el de discernir si el virus VS está evolucionando de una forma aleatoria o, por el contrario, se están seleccionando mutantes por un factor ambiental, como podría ser la inmunidad inducida por la vacuna en la población, dado que se han reportado datos que sugieren que VS podría infectar a vacunados, en especial por una exposición intensa al mismo, y el virus podría circular en poblaciones vacunadas seropositivas6,7.
CONCLUSIONES
La determinación de la secuencia de los genes de la Nucleoproteína y de la Hemaglutinina de aislados del virus del Sarampión ha permitido el identificar quince genotipos que circulan o han circulado a nivel mundial.
Existe una correspondencia entre los árboles filogenéticos obtenidos a partir de la secuencia del gen N y del gen H. Este resultado no indica que exista recombinación entre los distintos genotipos circulantes.
Aunque, en general, un genotipo no está restringido a un área geográfica, en cada area se pueden detectar genotipos locales. Algunos genotipos podrían haberse extinguido.
Tenemos evidencia de que un genotipo puede exportarse a partir de un caso a un área geográfica lejana (de Madrid a New Jersey, USA) y allí difundirse extensamente. Así, el análisis genético de aislados permite distinguir entre especies locales e importadas y evaluar la eficacia de las campañas de vacunación.
En una región geográfica pueden darse cambios bruscos en el genotipo circulante, como hemos detectado en Madrid en el otoño de 1993 y otros dos posibles en 1992 y 1970.
El cambio de genotipo circulante puede suponer variación en epítopos de la proteína H implicados en la neutralización del virus. Un cambio de aminoácido de la proteína H entre los genotipos C2 y D3 puede permitir la identificación de un nuevo epítopo implicado en la neutralización del virus.
En una serie temporal de aislados de un genotipo que apareció en Madrid, posiblemente importado, hemos podido evaluar, por primera vez, la velocidad de mutación de un gen del virus del Sarampión en la evolución natural de un genotipo a lo largo de una epidemia y para la proteína H hemos estimado 5x10-4 cambios por año para una posición de nucleótidos. Aunque es relativamente bajo, al menos diez veces menor del estimado para la proteína HN del virus de la gripe. A pesar de ser el virus del sarampión relativamente estable hemos observado la aparición de virus más mutados que no parecen haberse fijado.
BIBLIOGRAFIA
1. Griffin DE and Bellini W.J. Measles virus p 1267-1312. En: Fields B, et al eds Virology 3rd edition. New York: Lippincott-Raven Publishers; 1996. [ Links ]
2. Carabaña J [Tesis doctoral]. Madrid: Universidad Autónoma de Madrid; 1997. [ Links ]
3. Rima BK, Earle JAP, Yeo RP, Herlihy L, Baczko K, ter Meulen V, Carabaña J, Caballero M, Celma ML and Fernández-Muñoz R. Temporal and geographical distribution of measles virus genotypes, J Gen Virol 1995.76: 1173-1180. [ Links ]
4. Rima BK, Earle JAP, Baczko K, ter Meulen V, Liebert UG, Casterns C, Carabaña J, Caballero M, Celma ML and Fernández-Muñoz R. Sequence divergence of measles virus haemagglutinin during natural evolution and adaptation to cell culture. J Gen Virol 1998: 78: 97-106. [ Links ]
5. Rota JS, Heath JL, Rota PA, King GE, Celma ML, Carabaña R, Fernández-Muñoz R, Brown D, Jin L and Bellini WJ. Molecular Epidemiology of Measles Virus: Identification of pathways of transmission and implications for measles elimination. J Infect Dis 1998. 173: 32-37. [ Links ]
6. Paunio M, Peltola H, Valle M, Davidkin I, Virtanen m. and Heinonen O.P, Explosive school-based measles outbreak: Intense exposure have resulted in high risk, even among revaccinees. Am J Epidemiol 1998. 148: 1103-1110. [ Links ]
7. Damien B, Huiss S, Schneider F and Muller CP. Estimated susceptibility to asymptomatic secondary immune response against measles in late convalescent and vaccinated persons. J Med Virol 1998. 56: 85-90. [ Links ]

