










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkCuadernos de Medicina Forense
versión On-line ISSN 1988-611Xversión impresa ISSN 1135-7606
Cuad. med. forense vol.18 no.2 Málaga abr./jun. 2012
https://dx.doi.org/10.4321/S1135-76062012000200007
ATLAS DE PATOLOGÍA FORENSE
Bajo la dirección científica de la Sociedad Española de Patología Forense (SEPAF)
Muerte súbita por disección aórtica circunferencial con intususpección íntimo-intimal en un adulto joven con Síndrome de Noonan
Sudden death due to aortic circumferential dissection with intimo-intimal intussusception in a young adult with Noonan Syndrome
B. Cardona1, S. Giner1, J.M. Muñoz-Quirós1, P. Molina2, J.M. Ortiz3 y R. Bañón4
1Médico Forense. Servicio de Patología, Instituto de Medicina Legal de Alicante
2Especialista en Anatomía Patológica. Servicio de Patología, Instituto de Medicina Legal de Valencia
3Médico Forense. Servicio de Patología, Instituto de Medicina Legal de Valencia
4Médico Forense. Dirección del Instituto de Medicina Legal de Murcia
Dirección para correspondencia
RESUMEN
El Síndrome de Noonan es un trastorno genético de herencia autosómica dominante con expresividad variable y asociado a múltiples anormalidades. Las manifestaciones clínicas más importantes de este síndrome son entre otras la estatura corta, cuello ancho, hipertelorismo, exoftalmos y ptsosis palpebral. Suelen presentar malformaciones cardiacas tales como la displasia con estenosis valvular pulmonar y la miocardiopatía hipertrófica. El interés de este cuadro en Patología Forense se debe a que es posible su presentación en forma de muerte súbita sin ningún tipo de clínica previa. Se presenta el caso de una persona de sexo masculino de 18 años de edad, el cual se desplomó en la vía pública. Diagnosticado de Síndrome de Noonan sin controles médicos en ese momento. El estudio necrópsico reveló la presencia de una disección aórtica con intususpección íntimo-intimal.
Palabras clave: Síndrome de Noonan. Muerte súbita cardiaca. Válvula aórtica bicúspide. Patología Forense.
ABSTRACT
KNoonan syndrome is a relatively common autosomal dominant disorder whose pathophysiology is not fully understood. Its clinical features are characterized by dysmorphic facial features, webbing of the neck, proportionate short stature and heart disease (most commonly pulmonary valve stenosis and hypertrophic cardiomyopathy). In rare occasions, Noonan syndrome could have interest in forensic medicine due to their unexpected presentation as a sudden death episode without previous clinical symptoms. In this report, we present the case of a subject (male, 18 years old) who suddenly collapsed in the street. His parents known the previous NS diagnosis, but at the moment of death he was not under medical control or treatment. Autopsy findings revealed an aortic dissection with intimo-intimal intussusception.
Key words: Noonan syndrome. Sudden cardiac death. Bicuspid aortic valve. Forensic pathology.
Introducción
El síndrome de Noonan fue mencionado por primera vez en el año 1963 por los investigadores Noonan y Ehmke, cuando describieron nueve niños que presentaban anomalías cardíacas congénitas (estenosis pulmonar valvular) y un patrón característico de ojos oblicuos ampliamente espaciados con otras alteraciones de los rasgos faciales1.
Es un trastorno genético que se puede heredar de una forma autosómica dominante y que afecta al menos a 1 de cada 2.500 niños2. No obstante, una amplia mayoría de los casos parecen ser esporádicos (70%) y en todos ellos la mutación se ha producido en el alelo de origen paterno. La edad paterna parece incrementar el riesgo y, por causas desconocidas, el riesgo de presentar una mutación de novo es mayor en los hijos varones3. Sus principales características clínicas son: la talla baja (de inicio postnatal), el aspecto facial característico y los defectos cardíacos congénitos. La presentación clínica es similar a la del síndrome de Ullrich Turner, por eso también es conocido como "Síndrome de Pseudo Turner", "Síndrome de Turner del Varón" o "Síndrome de Turner masculino" y su gran confusión con éste ha llevado a un estudio minucioso de la genética del mismo, encontrándose un cariotipo normal a diferencia del síndrome de Turner (45X0).
En el síndrome de Noonan se han identificado 4 genes diferentes alterados: PTPN11, KRAS, RAF1 y SOS1; en aproximadamente el 50% de los casos existen mutaciones del gen PTPN114-9 en el cromosoma 12q24.1 y en un 20% de los casos, mutaciones en la vía RAS/MAPK (12p12.1)5 de señalización intracelular que opera en diversos procesos del desarrollo como la proliferación y diferenciación celular, organogénesis, plasticidad sináptica, crecimiento, apoptosis y envejecimiento celular3.
El fenotipo del síndrome de Noonan se caracteriza por estatura corta, cuello ancho, malformaciones del esternón con pectus carinatum (tórax en quilla) y excavatum (tórax en embudo), tórax ancho, mamas separadas y bajas, pterigiumcolli y pterigiumaxilar. El aspecto típico de la cara lo produce el hipertelorismo, el filtrum marcado (surco vertical en el centro del labio superior), las fisuras palpebrales antimongoloides, el paladar ojival, la micrognatía, las orejas displásicas de implantación baja y rotadas y los párpados gruesos con epicantus, exoftalmos y ptosis palpebral6. La talla final en estos pacientes se alcanza tardíamente y la media oscila entre 161-170±5 cm en los varones y entre 150-153 cm±5 cm en las mujeres (pérdida en la talla final de unas 2 desviaciones estándar respecto a la población de referencia)3. Los varones suelen tener criptorquidia y testículos pequeños1. Las malformaciones cardíacas más frecuentes son la displasia valvular pulmonar con estenosis variable y la miocardiopatía hipertrófica (MCH)7.
La muerte súbita (MS) en estos enfermos es extremadamente rara. Solo hemos encontrado tres casos descritos en la literatura: dos MS asociadas a MCH8 y una muerte súbita resucitada, también con MCH9.
Descripción del caso
Se trata de un varón de 18 años de edad, el cual se desplomó súbitamente en la vía pública. En sus antecedentes clínicos constaba estar diagnosticado del Síndrome de Noonan desde la infancia con desarrollo psicomotor normal y sin controles médicos en el momento del óbito. En la familia no existían otros miembros que tuvieran un fenotipo similar.
Hallazgos destacados en el examen externo: cadáver correspondiente a un varón de 175 cm de altura y 78 Kg de peso (IMC 25,5 kg/m2) con diversas características anatómicas destacadas: frente amplia, mentón prominente, pabellones auriculares de inserción baja y con lóbulos de las orejas hacia delante, ojos con hendiduras palpebrales hacia abajo, puente nasal grueso, cuello corto, deformidad esternal con pectus carinatum (tórax en quilla) prominencia del pubis y primer dedo de ambos pies en garra.
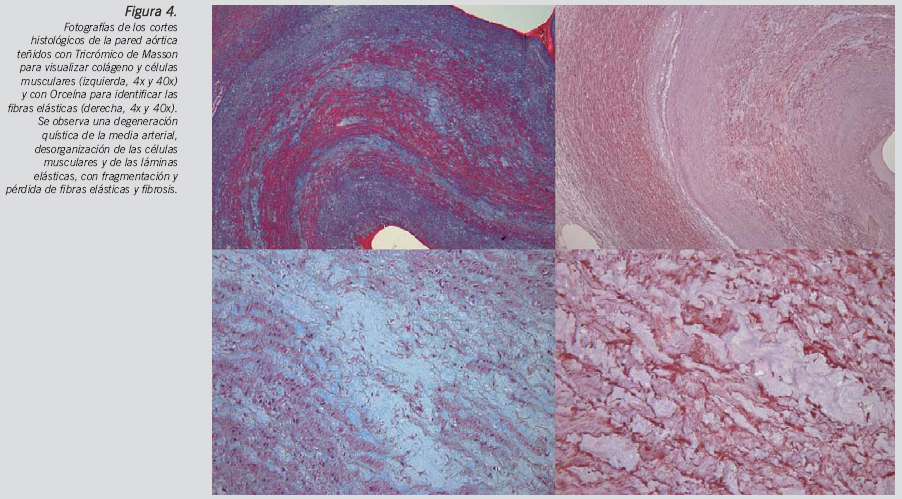
Hallazgos destacados en el examen interno: corazón de 395 g de peso (valor medio según peso corporal: 345 g [rango 261-455 g]). Grosor pared ventrículo izquierdo: 1,1cm, tabique interventricular: 1,1 cm y ventrículo derecho: 0,1 cm. Agujero oval no permeable. Válvulas aurículo-ventriculares y pulmonar normales. Válvula aórtica bicúspide. Severa dilatación de la raíz aórtica con disección circular completa de la íntima arterial a la altura del anillo de inserción valvular con desprendimiento completo e intususpección de 5 cm de longitud de la capa interna disecada hasta un área de menor calibre del cayado aórtico (Fig. 1 y 2), extendiéndose por la arteria subclavia izquierda y el tronco braquiocefálico, y obstruyendo completamente las tres luces vasculares (Fig. 3).



El estudio microscópico ofreció los siguientes datos de interés: signos moderados de hipertrofia miocitaria en miocardio ventricular izquierdo con patrón desorganizativo focal en tabique interventricular. Infiltración adiposa miocárdica ventricular derecha anterior y posterior con fibrosis intersticial sin reacción inflamatoria crónica. Necrosis quística de la media arterial aórtica (Fig. 4). Arterioloesclerosis renal con microlitiasis y esteatosis hepática en grado leve.
Discusión
Síndrome Noonan y patología cardiaca
El Síndrome de Noonan es un trastorno genético autosómico dominante, clínicamente heterogéneo y caracterizado por una estatura corta, dismorfia facial y torácica y un amplio espectro de alteraciones cardiacas congénitas10. El diagnóstico se basa en criterios clínicos, pero es complejo por la alta variabilidad de su expresión fenotípica. La afectación cardiaca está presente en el 50-80% de los afectados. Los defectos más frecuentemente encontrados son la estenosis pulmonar valvular (20-50%) y la miocardiopatía hipertrófica (20-30%). Se han descrito también, aunque en menor proporción, defectos septales atriales (6-10%) y ventriculares (5%) o ductus arterioso persistente (3%)11. Existen muy pocos casos descritos de síndrome de Noonan con patología valvular aórtica: tres casos con estenosis valvular12-14 y uno con válvula bicúspide15 y/o dilatación de la raíz aórtica16 y en sólo dos de ellos complicada con disección de la pared arterial17,18. La presencia de un corazón normal en el período neonatal o en la primera infancia no descarta la posibilidad de que estos pacientes desarrollen una cardiopatía más tardíamente, por ello deben realizarse evaluaciones cardiológicas periódicamente a lo largo de la infancia y adolescencia3. La dilatación aórtica no es una lesión típica del Síndrome de Noonan, aunque puede estar infradiagnosticada16. Este hecho justifica la importancia del seguimiento a largo plazo de estos enfermos con medición de diámetros aórticos.
Válvula aórtica bicúspide y aortopatía
La válvula aórtica bicúspide es la anomalía cardiaca congénita más frecuente, con una prevalencia estimada de 0,5-2% y una mayor afectación del sexo masculino (3:1)19. La válvula aórtica bicúspide puede producir estenosis o regurgitación, sin embargo, el diámetro aumentado de la raíz de la aorta que ocurre en más de la mitad de los casos es independiente de alteraciones hemodinámicas o de la edad20. En la aorta ascendente de estos pacientes se ha demostrado un flujo aórtico característicamente diferente y un sustrato anatómico que justifica la dilatación de la aorta y sus complicaciones, caracterizado por lesiones histológicas de la capa media no proporcionales a la severidad de la lesión asociada21. Esta serie de cambios histopatológicos degenerativos de la capa media incluyen la necrosis quística, una apoptosis aumentada y alteración de la orientación en las células musculares y un menor desarrollo de las fibras elásticas con fragmentación de las mismas y menor contenido en fibrilina 1, que provocan una debilidad y dilatación progresiva de la pared de la aorta con el consiguiente riesgo aumentado de disección22-24. Por otro lado, se ha sugerido un defecto común del desarrollo de la válvula aórtica y la pared de la aorta, apoyado en su demostración experimental25 y en el origen neuroectodérmico de ambas estructuras26. Todo ello ha inducido a no considerar a la válvula aortica bicúspide como un hallazgo aislado, sino formando parte de un mayor espectro patológico de anormalidades de la pared de la aorta que podrían englobarse bajo un mismo síndrome19. Sin embargo, el patrón de flujo posvalvular no axial y turbulento dirigido hacia la pared anterolateral de aorta y la demostración de las alteraciones histológicas de distribución excéntrica localizadas únicamente en la convexidad de la aorta ascendente y arco aórtico, irían en contra de una teoría patogénica exclusivamente genética y mantienen la actual controversia en este tema21.
El riesgo de disección de la aorta en la válvula aórtica bicúspide es mayor, siendo 5-9 veces más frecuente que en la población general19. No obstante, las dimensiones de la aorta no constituyen un buen indicador ya que las alteraciones estructurales que debilitan la pared de la aorta pueden producirse sin dilatación21,27.
Disección aórtica circunferencial
Por otro lado, la disección aórtica circunferencial con intususpección íntimo-intimal es una forma rara de presentación de la disección tipo A de Standford (tipo II de Bakey), con muy pocos casos publicados. Los primeros casos descritos datan de 188728 y 190929 y se describen como "inversión del cilindro interno"30. Se produce un desgarro completo de la intima con invaginación de la misma pudiendo producir obstrucción de la luz aórtica, de las ramas supraaórticas (en cuyo caso se asocia sintomatología neurológica) o del ventrículo izquierdo y orificios coronarios31,32. El diagnóstico es complicado necesitándose un alto índice de sospecha. El tratamiento quirúrgico, si es posible, permite su reparación completa31-33.
En este caso, la ectasia de la raíz aórtica provocó la obstrucción completa de la aorta, por el segmento invaginado en su tramo no dilatado, así como la obstrucción de los troncos supraaórticos, provocando la muerte inmediata del paciente.
Bibliografía
1. Pacheco LM, Sánchez MA, Sánchez DL, Rosales J. Síndrome de Noonan, presentación de un caso. Medisan 2002;6(1):86-90. [ Links ]
2. Tobar LN, Mejía LJ. Síndrome de Noonan. Acta de Otorrinolaringología & Cirugía de Cabeza y Cuello 2004;32(4):131-4. [ Links ]
3. Pozo J. Síndrome de Noonan y síndromes relacionados (síndromes neuro-facio-cardio-cutáneos). Pediatr Integral 2010;XIV(8):636-47. [ Links ]
4. Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 2001;29(4):465-8. [ Links ]
5. Ezquieta B, Santomé JL, Carcavilla A, Guillén-Navarro E, Pérez-Aytés A, et al. Alterations in RAS-MAPK genes in 200 Spanish patients with Noonan and other neuro-cardio-facio-cutaneous syndromes. Genotype and cardiopathy. Rev Esp Cardiol 2012;65(5):447-55. [ Links ]
6. Limal JM, Bonnet D, Bouc YL, Leheup B, Lyonnet S. Noonan syndrome: an enigma. Arch Pediatr 1998;5(7):715-8. [ Links ]
7. Limongelli G, Hawkes L, Calabro R, McKenna WJ, Syrris P. Mutation screening of the PTPN11 gene in hypertrophic cardiomyopathy. Eur J Med Genet 2006;49(5):426-30. [ Links ]
8. Ishizawa A, Oho S, Dodo H, Katori T, Homma SI. Cardiovascular abnormalities in Noonan syndrome: The clinical findings and treatments. Acta Paediatr Jpn 1996;38(1):84-90. [ Links ]
9. Aydin A, Yilmazer MS, Gurol T. Sudden death in a patient with Noonan syndrome. Cardiol Young 2011;21(2):233-4. [ Links ]
10. Noonan JA. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am J Dis Child 1968;116(4):373-80. [ Links ]
11. Van der Burgt I. Noonan syndrome. Orphanet J Rare Dis 2007;2:4. [ Links ]
12. Abadir S, Edouard T, Julia S. Severe aortic valvar stenosis in familial Noonan syndrome with mutation of the PTPN11 gene. Cardiol Young 2007;17(1):95-7. [ Links ]
13. Danetz JS, Donofrio MT, Embrey RP. Multiple left-sided cardiac lesions. Cardiol Young 1999;9:610-2. [ Links ]
14. Mollica N, Antoncecchi S, Antoncecchi Jr E, et al. Combined aortic and pulmonary valve stenosis, changes in primary hemostasis and partial deficiency of factor XII in Noonan's syndrome. Minerva Cardioangiol 1987;35:311-5. [ Links ]
15. Petersen SE, Mohrs OK, Theile U, Pohlmann W, Voigtlander T. Noonan's syndrome with an unusual combination of hypertrophic cardiomyopathy, congenital bicuspid aortic valve, coarctation of the aorta and hypoplastic aortic arch. Z Kardiol 2004;93(4):310-6. [ Links ]
16. Power PD, Lewin MB, NC Hannibal, MC Glass. Aortic Root Dilatation is a Rare Complication of Noonan Syndrome. Pediatr Cardiol 2006;27:478-480. [ Links ]
17. Kretschmar K, Witkowski R. Dissecting aortic aneurysm in a man with symptoms of Turner's síndrome. Z Gesamte Inn Med 1982;37(9):278-81. [ Links ]
18. Shachter N, Perloff JK, Mulder DG. Aortic dissection in Noonan's syndrome (46 XY turner). Am J Cardiol 1984;54(3):464-5. [ Links ]
19. Sorrell VL, Pancyzk E, Alpert JS. A new disease: bicuspid aortic valve aortopathy syndrome. Am J Med 2012;125(4):322-3. [ Links ]
20. Ward C. Clinical significance of the bicuspid aortic valve. Heart 2000;83: 81-85. [ Links ]
21. Gallego P. Enfermedad vascular de la pared arterial en las cardiopatías congénitas. Rev Esp Cardiol Supl 2009;9:30E-39E. [ Links ]
22. Plaisance BR, Winkler MA, Attili AK, Sorrel VL. Congential bicuspid aortic valve first presenting as an aortic aneurysm. Am J Med 2012;125:e5-e7. [ Links ]
23. Bonderman D, Gharehbeghi-Schnell E, Wollenek G, et al. Mechanism underlying aortic dilatation in congenital aortic valve malformation. Circulation 1999. 99:2138-43. [ Links ]
24. Niwa K, Perloff JK, Bhuta SM, Laks H, Drinkwater DC, Child JS, et al. Structural abnormalities of the great arterial walls in congenital heart disease. Circulation 2001;103:393-400. [ Links ]
25. Kirby ML, Turnage KD, Hays BM. Characterization of conotruncal malformations following ablation of "cardiac" neural crest. Anat Rec 1985;213:87-93 [ Links ]
26. Kirby ML, Waldo KL. Role of neural crest in congenital heart disease. Circulation 1990;82:332-40 [ Links ]
27. Basso C, Nistri S, Rizzo S, Abudureheman A, Thiene G. Bicuspid aortic valve, aortic dissection and sudden death in the young: just a matter of ascending aorta diameter? Circulation 2007;116:II463. [ Links ]
28. Bostroem E. Das geheilteaneurysmadissecans. Deut Arch Klin Med 1887;42:1. [ Links ]
29. Chiari H. Aneurysma dissecans aortae mit inversion des inneren zylinders. Verh Deut Path Ges 1909;207. [ Links ]
30. Touati G, Carmi D, Trojette F, Jarry G. Intimo-intimal intussusception: a rare clinical form of aortic dissection. Eur J Cardiothorac Surg 2003 Jan;23(1):119-21. [ Links ]
31. Lajevardi SS, Sian K, Ward M, Marshman D. Circumferential intimal tear in type A aortic dissection with intimo-intimal intussusception into left ventricle and left main coronary artery occlusion. J Thorac Cardiovasc 2012 Jun 5. (Epub ahead of print). [ Links ]
32. Yamabi H, Imanaka K, Sato H, Matsuoka T. Extremely localized aortic dissection and intussusception of the intimal flap into the left ventricle. Ann Thorac Cardiovasc Surg 2011;17(4):431-3. [ Links ]
33. Niino T, Unosawa S, Shimura K. Stanford type A aortic dissection with intimal intussusception. Gen Thorac Cardiovasc Surg. 2012 May 22. (Epub ahead of print). [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Begoña Cardona Valencia
Servicio de Patología
Instituto de Medicina Legal
Avenida Aguilera, S/N
03006 Alicante
E-mail: cardona_beg@gva.es
Fecha de recepción: 12.JUN.2012
Fecha de aceptación: 18.JUN.2012

