










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versão impressa ISSN 1137-6627
Anales Sis San Navarra vol.26 supl.1 Pamplona 2003
Intoxicaciones medicamentosas (II). Analgésicos y anticonvulsivantes
Acute pharmacologic poisoning (II). Analgesics and anticonvulsants
P. Munné1, J.J. Saenz Bañuelos2, J.J. Izura2, G. Burillo-Putze3, S. Nogué1
| RESUMEN En este segundo capítulo sobre Intoxicaciones Medicamentosas Agudas abordamos dos grupos de sustancias de enorme trascendencia desde el punto de vista de su uso y morbimortalidad. Dentro del grupo de los anagésicos-antiinflamatorios desarrollamos el paracetamol y los salicilatos, de enorme disponibilidad para la población. En cuanto a los anticonvulsivantes, aunque están poco implicados en el conjunto de las intoxicaciones medicamentosas agudas, sus efectos pueden ser graves. Nos ceñimos a cuatro fármacos: ácido valproico, fenobarbital, carbamacepina, y fenitoína. Finalmente dedicamos un apartado a la isoniacida, fármaco que, con el rebrote de la tuberculosis, presenta interés toxicológico. | ABSTRACT In this second chapter on Acute Drugs Poisoning we deal with two groups of substances of great transcendence from the point of view of their use and morbidity/mortality. Within the group of analgesic-anti-inflammatory drugs we consider paracetamol and the salicylates, which are easily available to the population. With respect to the anticonvulsants, although they are barely involved in the ensemble of acute drug poisonings, their effects can be serious. We concentrate on four drugs: valproic acid, phenobarbitol, carbamacepine, and phenytoin. Finally, a section is dedicated to isoniazid, a drug that, with the renewed incidence of tuberculosis, is of toxicological interest. |
| 1. Unidad de Toxicología Clínica. Hospital Clínic. Barcelona. 2. Servicio de Medicina Intensiva. Hospital Virgen del Camino. Pamplona. 3. Servicio de Urgencias. Hospital Universitario de Canarias. Tenerife. | Correspondencia: Pere Munné Mas Servei d´Urgencias Unitat de Toxicologia Clinica Hospital Clinic C/ Villarroel 170 08036 Barcelona Tel. 93 2279833, 93 2275400 ext.2666 Fax 93 2275693 e-mail: jmilla@medicina.ub.es |
PARACETAMOL
El paracetamol o acetominofen es un n-acetil-p-aminofenol derivado de la fenacetina, de bajo peso molecular (151 dalton) que se comporta como un ácido débil. Se encuentra comercializado en forma de cápsulas o comprimidos, conteniendo cada uno 500, 650 ó hasta 1000 mg. Existen también presentaciones en gotas (100 mg /ml) y en solución. Hay preparados con codeína en distintas proporciones y otros, en general antigripales, que además de paracetamol, contienen diversos fármacos (antihistamínicos, cafeína, efedrina, ascórbico, analgésicos, etc.). Su toxicidad deriva habitualmente de la presencia de paracetamol aunque debe siempre valorarse la acción narcótica de la codeína.
En la actualidad es el analgésico-antipirético de mayor uso, especialmente en pediatría para evitar la asociación salicílicos-síndrome de Reye. Esta mayor accesibilidad ha condicionado un incremento en el número de intoxicaciones agudas por paracetamol hasta convertirse en la primera causa de intoxicación medicamentosa en pediatría1 y ocupar los primeros lugares en la casuística de adultos tras las benzodiacepinas y los antidepresivos2.
Cinética
El paracetamol se absorbe por vía digestiva de forma rápida alcanzando un pico plasmático a los 40-60 minutos (30 minutos en preparados líquidos)3. En sobredosis la mayor parte de paracetamol se absorbe en 2 horas pero no alcanza el pico plasmático hasta las 4 horas, aunque puede ser a las 6 ó más horas. Ello acontece con mayor frecuencia si hay ingesta simultánea de opiáceos (codeína) o anticolinérgicos, en preparaciones galénicas retard (no disponibles en nuestro medio) o si hay presencia de comida en cavidad gástrica, especialmente rica en carbohidratos4.
La biodisponibilidad es alta (60-95%). La unión a proteínas es de 10-30 % pudiendo alcanzar el 50% en sobredosis. El volumen de distribución es de 0,8-1 l/Kg5. Atraviesa la placenta y la barrera hematoencefálica. La leche contiene menos de un 2% de la dosis materna.
La vía principal de eliminación (más del 90% de la cantidad) es la biotransformación hepática a través de tres mecanismos metabólicos6: la glucoroconjugación (40-67%) por acción de la glucoroniltrasferasa, la sulfoconjugación (20-46%, más en niños) mediada por la sulfotransferasa y una vía menor oxidativa, (usualmente entre el 5-15%) a través del sistema citocromo P450. La glucuro y la sulfoconjugación son procesos saturables que generan metabolitos atóxicos que se eliminan por la orina.
La vía oxidativa produce un metabolito reactivo altamente tóxico que en condiciones terapéuticas se une con el glutation celular formándose conjugados de cisteína y ácido mercaptúrico.
En sobredosis, al saturarse las vías metabólicas principales, una mayor proporción de paracetamol se biotransforma a través del sistema oxidativo generándose una mayor cantidad de metabolito tóxico (n-acetil-benzo-quinoneimina o NAPQI). Esta mayor producción de NAPQI excede la capacidad desintoxicante del glutation. Se cree que el NAPQI no neutralizado es el máximo responsable de la acción tóxica hepática y renal.
La vida media del paracetamol es de 1-3 horas7 prolongándose en neonatos, ancianos y cirróticos. En casos de sobredosis puede alcanzar las 12 horas.
Mecanismo de acción
El paracetamol no es tóxico. Su toxicidad es debida a la acción del metabolito intermedio (NAPQI) generado al biotrasnformarse a través de la vía oxidativa hepática. A dosis terapéuticas, el NAPQI generado se une al glutation intracelular y a otros compuestos tiólicos formándose un conjugado atóxico tal como se ha mencionado en el apartado anterior.
En sobredosis, cuando la cantidad de paracetamol supera una dosis crítica (generalmente 150 mg/kg8), las vías de glucuro y sulfoconjugación se saturan incrementándose la proporción de paracetamol que seguirá la vía oxidativa. Ello aumenta la velocidad y la producción de NAPQI precisándose más gutation para neutralizarlo9.
Cuando las reservas de glutation hepático descienden por debajo de un 30%10, el NAPQI libre ejerce su acción tóxica sobre el hepatocito uniéndose mediante un enlace covalente al locus neutrofílico de determinadas proteínas intracelulares, pudiendo producir la muerte celular. La necrosis inicialmente se concentra en la zona III centrolobulillar ya que es aquí donde hay un mayor metabolismo oxidativo extendiéndose al restante parénquima hepático en los casos más severos.
Un mecanismo de acción similar (formación de NAPQI a través del P450 renal) se ha sugerido como causa de la necrosis tubular que ocasionalmente también acaece en esta intoxicación11.
Aparte de este mecanismo oxidativo, se han propuesto experimentalmente otras vías fisiopatológicas complementarias o, incluso, determinantes del daño hepático y extrahepático: formación de radicales libres12, cambios isquémicos en la microcirculación13, trastornos de la homeostasis cálcica14, inhibición de la cadena respiratoria mitocondrial15, etc.
Manifestaciones clínicas
La intoxicación aguda por paracetamol presenta tres características:
1. El órgano diana es el hígado. También pueden afectarse otros órganos aunque muy raramente.
2. La clínica inicial (primeras 12-24 h) es, en general, asintomática o leve incluso en los casos que desarrollarán después una alta toxicidad.
3. Es una intoxicación potencialmente muy grave, incluso letal.
Clásicamente el cuadro clínico cursa en tres períodos bien definidos pudiendo añadirse un cuarto período resolutivo en los casos en que se logra la curación.
Período inicial (0-24 h)
En general, el paciente está asintomático aunque puede cursar con náuseas, vómitos, epigastralgia, malestar, palidez y diaforesis. La concentración de aspartato aminotransferasa (AST o GOT más sensible que la ALT o GPT) se mantiene generalmente normal durante la mayor parte de este período, aunque se puede observar con frecuencia en el último tercio de estas 24 horas iniciales una citolisis incipiente. Aunque muy inusual pueden haber AST elevadas al cabo de 8-12 h en caso de sobredosis muy copiosas.
A pesar de la normalidad de las transaminasas, se pueden observar en este período disminuciones moderadas del quick, en ausencia de hepatotoxicidad y sin mayor trascendencia clínica ni evolutiva16. Ocasionalmente también puede observarse hiperglicemia17.
Excepcionalmente, se han descrito casos de acidosis metabólica y coma que aparecen a las 4-6 horas (hasta 12 h) después de ingestas masivas (75 o más g de paracetamol con niveles séricos de 800 µg / ml) en ausencia de hepatotoxicidad18.
Sólo una parte de los pacientes pasan el período siguiente. El resto evoluciona favorablemente sin desarrollar toxicidad hepática.
Período segundo o intermedio (24- 72 h)
Clínicamente el enfermo puede seguir asintomático (lo más frecuente), presentar una sintomatología leve, similar a la del primer período y/o iniciar un dolorimiento en hipocondrio derecho.
La alteración característica de este período es el aumento de los enzimas hepáticos (GOT y GPT) que puede iniciarse a las 24 h post-ingesta, siendo más habitual y casi general (en los pacientes que han evolucionado a este período) a las 36 h, alcanzando su nivel máximo a los 3-4 días. Esta elevación enzimática puede acompañarse de una sintomatología de hepatitis infecciosa. En este período también pueden alterarse otros parámetros (bilirrubina, quick, antitrombina III) según sea la gravedad de la intoxicación.
Básicamente este estadio expresa el grado de lesión-necrosis hepática pudiendo evolucionar hacia la normalidad en muy pocos días o bien entrar en la fase siguiente de fracaso de la función hepática.
Aparte de la hepatotoxicidad, de forma inusual pueden afectarse otros órganos: riñón, miocardio, páncreas y excepcionalmente sistema vascular (hipotensión, shock18) y sistema hematológico (trombocitopenia, pancitopenia18, nunca metahemoglobinemia a pesar de ser el paracetamol un derivado fenacetínico).
La insuficiencia renal aguda (necrosis tubular) es infrecuente (0,4-4%) y casi siempre se asocia con hepatotoxicidad severa aunque también puede presentarse sin participación hepática19. En general aparece a los 2-5 días post-ingesta.
Algo más común es el trastorno moderado de la función renal aparejado al fallo hepático agudo. El dolor lumbar y la presencia de proteinuria y/o hematuria pueden ser precursores del fallo renal20.
Aparte de la afectación hepática y renal, las alteraciones miocárdicas (depresión ST, T invertida, aumento CKMB) o pancreáticas (más hiperamilasemia que clínica) son muy inusuales y siempre aparecen como manifestaciones acompañantes de la hepatotoxicidad.
Período tercero (72h- 5 días)
Es el período de la máxima expresión de hepatotoxicidad pudiendo evolucionar a fallo hepático agudo. La clínica puede variar desde ser poco expresiva a desarrollar encefalopatía, coma y/o trastornos de coagulación según el grado de disfunción hepática.
Las GOT-GPT alcanzan valores máximos aunque las alteraciones del INR, la bilirrubinemia, la hipoglicemia y la acidosis metabólica definen mejor el grado de insuficiencia hepática.
La evolución puede ser aún favorable con tratamiento (terapia de soporte, N-acetilcisteína, transplante) o incluso sin él.
El éxitus (5-7 días) por fallo hepático agudo sobreviene por alteración multiorgánica incluyendo insuficiencia renal, hemorragia, distrés respiratorio, sepsis y, básicamente, edema cerebral.
Período último o curativo (5-7 días hasta 2 semanas)
Si la insuficiencia hepática aguda no acaba en exitus, la reversibilidad es total, regenerándose por completo todo el tejido hepático.
Factores que pueden modificar la hepatotoxicidad del paracetamol
Hepatopatía crónica
No hay evidencia de que una hepatopatía crónica estable aumente la hepatotoxicidad del paracetamol a dosis terapéuticas y, probablemente, tampoco en sobredosis.
Alcoholismo crónico
El paracetamol a dosis terapéuticas no parece que cause hepatotoxicidad en alcohólicos crónicos21. En cambio, hay datos que avalan una mayor toxicidad hepática en caso de intoxicación aguda en pacientes enólicos crónicos22.
Ingesta simultánea de alcohol
Si se produce una ingesta enólica aguda simultánea con la sobreingesta, el alcohol es probable que disminuya la capacidad tóxico-hepática del paracetamol, en especial, en enólicos crónicos.
Esta protección desaparece si el tiempo entre la ingesta de alcohol y la sobredosis de paracetamol supera las 8 horas23.
Casos pediátricos
Después de una sobredosis (en ingesta única) de paracetamol, los niños menores de 6 años, desarrollarán una menor hepatotoxicidad que los adolescentes y adultos en condiciones similares de dosis y tratamiento24.
Tratamiento crónico con otros fármacos
La terapéutica con isoniazida25 y con anticonvulsivantes (excepto, probablemente barbitúricos y fenitoína21) incrementa la toxicidad hepática del paracetamol en sobredosis.
Enfermedades previas
Cuando el paracetamol se toma a dosis terapéuticas o supraterapéuticas pero subtóxicas y con un régimen de dosificación repetida y continuada, varios estudios sugieren que en tales casos puede producirse un aumento de la hepatotoxicidad si coexisten determinadas patologías previas tales como: enfermedad de Gilbert, desnutrición severa y HIV evolucionado.
En la intoxicación aguda, hay datos que también avalan esta potenciación de la toxicidad del paracetamol en pacientes con las enfermedades anteriormente citadas.
Diagnóstico: analítica toxicológica
El análisis de la concentración sérica de paracetamol es la exploración básica para confirmar el diagnóstico. El resultado no sólo tiene valor de certeza diagnóstica sino que, evalúa el riesgo de hepatotoxicidad indicando si debe administrarse o no el antídoto específico. Para ello se utiliza el Nomograma de Rumack-Mathew (como se verá en el apartado de tratamiento) que indica si debe administrarse el antídoto según la concentración plasmática de paracetamol en relación con el intervalo transcurrido desde la ingesta del tóxico.
El análisis toxicológico tiene valor diagnóstico y pronóstico si se realiza entre las 4 horas y las 24 horas post-ingesta. Antes de las 4 h continúa teniendo valor para el diagnóstico pero carece de valor pronóstico ya que la absorción no es completa.
En el extremo opuesto, transcurridas 24 horas, el paracetamol se habrá metabolizado por lo que será indetectable en la analítica, no pudiendo negar ni confirmar ningún diagnóstico.
En cuanto al perfil hepático, aunque su posible alteración por efecto del paracetamol se produce más tardíamente, debe solicitarse siempre en la valoración inicial para disponer de resultados basales que servirán de comparación en valoraciones posteriores.
La elevación de las SGOT y/o SGPT que se produce entre las 24-48 h post-ingesta en los casos que cursan con hepatotoxicidad, pueden confirmar el diagnóstico en las intoxicaciones atendidas transcurridas más de 24 h donde el paracetamol sérico, como ya se ha mencionado, no es de utilidad al ser indetectable.
Durante el curso evolutivo, además de las transaminasas se solicitarán los restantes parámetros de función hepática (glicemia, quick, bilirrubina, equilibrio ácido-base) no como ayuda diagnóstica sino como indicadores de gravedad.
Tratamiento
Ante una intoxicación aguda por paracetamol deberán evaluarse la puesta en práctica de los siguientes aspectos terapéuticos:
1. Descontaminación digestiva
2. Antídoto específico (N-acetilcisteína o NAC).
3. Terapia de soporte
Descontaminación digestiva
Se ha postulado que en esta intoxicación, las indicaciones de la descontaminación digestiva eran limitadas por diversas razones: la rápida absorción del paracetamol, el disponer de un antídoto eficaz que relega a un segundo plano las medidas para disminuir la absorción y, por último, que la eficacia del antídoto si se administra per os, podría reducirse por la acción de la ipeca o del carbón activado. En nuestro medio, debido a que es más frecuente la utilización de la NAC por vía endovenosa se puede emplear cualquier método de descontaminación digestiva (ipeca, carbón, lavado) sin las objeciones anteriormente citadas.
No obstante, la mayoría de autores, prefieren usar carbón activado ya que es el único método descontaminante en que se ha comprobado que su administración decrece el número de pacientes que precisan tratamiento antidótico26, por otra parte absorbe muy bien al paracetamol27 y por último, incluso en el supuesto de utilizar NAC por vía oral, la posible adsorción del NAC por parte del carbón activado carece, al parecer, de significado clínico28.
La dosis óptima de carbón activado no está totalmente establecida aunque puede utilizarse la pauta habitualmente aceptada: 25 a 100 g en adultos y adolescentes, 25-50 g en niños de 1 a 12 años y 1 g/Kg en menores de un año.
En sobreingestas de paracetamol muy elevadas, consecuentemente se aumentará la dosis de carbón hasta alcanzar el límite alto anteriormente expuesto. No es inusual que se presenten vómitos tras la administración del carbón activado. En tal caso, debe repetirse la dosis adjuntando un antiemético. Se practicará la descontaminación digestiva preferentemente con carbón activado en las siguientes circunstancias:
– Si el tiempo transcurrido desde la ingesta es menor de 2 horas (aunque es más eficaz dentro de la 1ª hora).
– Si la cantidad de paracetamol presuntamente alcanza la dosis considerada tóxica: > 125-150 mg/Kg en adultos y adolescentes o > 200 mg/Kg en niños menores de 6 años29.
– En el caso de que la cantidad ingerida no sea razonablemente fiable o no pueda averiguarse, a efectos de la descontaminación digestiva, deberá considerarse que está dentro del dintel tóxico descrito.
– En caso de intoxicación mixta, con independencia del paracetamol, la descontaminación digestiva estará indicada siempre que el tóxico acompañante lo requiera.
Antídoto específico
La N-acetilcisteína o NAC es el antídoto específico que actúa a través de varios mecanismos: incrementando la síntesis de glutation30, uniéndose al NAPQI, formando conjugados atóxicos31 y aumentando la vía metabólica de la sulfoconjugación32. Se cree además que el NAC también tiene una acción no específica antioxidante que preserva el fallo multiorgánico en caso de insuficiencia hepática aguda.
La eficacia de la NAC es máxima si se administra dentro de las primeras 8 horas de la intoxicación. Este período es el tiempo aproximado en que se deplecciona un 70% del glutation hepático al conjugarse con el NAPQI.
El 30% restante es ya insuficiente para unirse a la totalidad del NAPQI que se va formando. El NAPQI libre comienza su acción hepatotóxica transcurridas estas 8 horas que la NAC no podrá ya impedir. En los casos atendidos después de este intervalo de 8 horas el grado de hepatoprotección de la NAC irá decreciendo proporcionalmente al intervalo asistencial. La indicación para administrar el antídoto se basa en dos parámetros principales: la cantidad de paracetamol ingerido y el tiempo transcurrido desde su ingesta.
Debido a que el conocimiento de la cantidad de tóxico que se ingirió presenta una fiabilidad irregular y debido a las posibles diferencias entre lo ingerido y lo absorbido, en la práctica, como parámetro de cantidad de tóxico absorbido se utiliza la concentración sérica de paracetamol.
Analizando retrospectivamente los casos que presentaron hepatotoxicidad (SGOT > 1000 UI/l) respecto a los que no la presentaron, y en base a la vida media del paracetamol, Rumack y Mattehew en 197533 publicaron su famoso nomograma donde se puede predecir el riesgo de hepatotoxicidad (y por lo tanto la indicación de administrar NAC) en función de los dos parámetros ya mencionados: concentración sérica de paracetamol e intervalo transcurrido entre la ingesta y la obtención de la muestra (Fig.1).

El nomograma sólo es válido cuando la concentración sérica de paracetamol se obtiene entre las 4 y las 24 horas después de una sobredosis aguda (dosis única). No es utilizable si se trata de una ingesta crónica o fraccionada.
En el nomograma original la línea de separación entre riesgo y no riesgo de hepatotoxicidad empezaba con una concentración plasmática de paracetamol de 200 µg/ml a las 4 horas de la ingesta, continuaba a las 12 horas postingesta con valores de 50 µg/ml de paracetamol (considerando una vida media de 4 h) y finalizaba a las 24 horas con paracetamolemias del orden de 6,25 µg/ml. Este nomograma 200 continua vigente en algunos lugares especialmente el Reino Unido tratándose con NAC los casos con niveles de paracetamol por encima de la línea 200.
Se han publicado casos que presentaron hepatotoxicidad grave a pesar de tener niveles de paracetamol sin riesgo (por debajo de la línea 200) y que, consecuentemente, no fueron tratados con NAC. Aunque estos fallos de nomograma se produjeron en algunos pacientes con factores predisponentes a una mayor toxicidad hepática, se han referido también casos sin presentar estos factores de riesgo34. Debido a ello y a la experiencia acumulada durante más de 20 años, de forma arbitraria, se redujeron un 25% los valores de las paracetamolemias del nomograma original. De esta forma se creó el nomograma 150 más conservador, cuyo desarrollo en cifras se detalla en la tabla 1.

El nomograma 150 es hoy el más utilizado para poder indicar o no el tratamiento con NAC. Su especificidad y fiabilidad es muy alta por lo que si se aplica adecuadamente (analítica correcta y tiempo post-ingesta conocido) es de una gran seguridad en la indicación del tratamiento con NAC.
Los casos con riesgo hepatotóxico aumentado (desnutrición, VIH, inducción enzimática, isoniazida), en general también se les puede aplicar el nomograma 150 con total validez aunque en los casos dudosos o graves pueden reducirse los valores de paracetamolemia del nomograma 150 en un 25% adicional para mayor seguridad.
En el Anexo 1 se presentan los criterios para la indicación de NAC en las diferentes situaciones clínicas. Existen dos pautas básicas de administración de la NAC: la vía oral y la vía parenteral. Cada una tiene una posología y una duración distintas (Tabla 2).

Abogamos por emplear la pauta de administración intravenosa en base a los siguientes razonamientos:
– La eficacia de la pauta oral respecto a la i.v. es idéntica si se trata al paciente antes de las 8-10 horas post-ingesta.
– La pauta oral (72 h, 1330 mg/Kg de NAC) es más eficaz que la pauta i.v. (20 h, 300 mg/Kg de NAC en los casos tratados tardíamente, tanto para prevenir la toxicidad hepática como para tratarla si ya se hubiese desarrollado35. Probablemente ello se debe a la mayor aportación de NAC en la pauta oral y la mayor duración del tratamiento. Debido a ello, es necesario prolongar el tratamiento con NAC i.v. en los casos tratados tardíamente si desarrollan citolisis severa o insuficiencia hepática.
– Esta mayor duración y aporte de NAC i.v. logra también mejorar el pronóstico de la hepatotoxicidad36.
– La pauta oral provoca vómitos con frecuencia, necesitando administrar antieméticos y dosis adicionales, lo que no asegura la cantidad de NAC que finalmente se absorbe.
– Los efectos adversos producidos por la NAC i.v. incluyen eritema cutáneo, prurito, náuseas, vómitos, angioedema, broncoespasmo, anafilaxia y excepcionalmente, éxitus. Con menor frecuencia pueden aparecer taquicardia, hipotensión o hipertensión. La mayoría de casos se resuelven enlenteciendo o suspendiendo la perfusión de NAC y administrando un antihistamínico i.v. Los efectos adversos graves prácticamente han desaparecido al administrar la dosis inicial (150 mg/Kg) en una hora en vez de los 15 minutos que se recomendaban. Con esta menor velocidad de perfusión el perfil de seguridad es relativamente igual entre las dos pautas oral e intravenosa.
– La pauta oral puede colisionar con la eficacia de la descontaminación digestiva (la NAC puede reducir la capacidad adsortiva del carbón activado sobre el paracetamol37), aunque no se ha probado que ello tenga relevancia clínica. Por otra parte, la emesis y el carbón activado pueden reducir la cantidad de NAC absorbida38 aunque otros autores lo niegan39 precisando, en tal caso, aumentar la dosis de NAC.
En cuanto a la duración del tratamiento con NAC, aunque existen algunas pautas de administración de NAC distintas de las dos clásicas (oral 72 h y parenteral 20 h.) nos ceñiremos a estas dos para exponer la duración adecuada del tratamiento con NAC.
Cuando se use la pauta intravenosa (21 horas), debe administrarse la totalidad del tratamiento. No está indicado suspenderlo al comprobar que la paracetamolemia se ha negativizado. Debe prolongarse (150 mg/Kg/24 h), como ya se ha expuesto, si aparecen signos de citolisis o insuficiencia hepática especialmente en los casos con intervalo asistencial superior a 10 horas.
En cuanto a la pauta oral (72 horas) también hay que completar todo el tratamiento. Puede suspenderse sin perder eficacia antidótica si a las 24 h el paracetamol es indetectable con GOT/GPT normales40. Otro estudio establece que puede acortarse la duración del tratamiento hasta las 36 h de iniciado, sin perder eficacia siempre que las GOT/GPT se mantengan en valores de referencia41.
Terapéutica de soporte
La persistencia de náuseas y vómitos puede precisar algún antiemético (metoclopramida) pudiendo utilizarse ondasentrón (quizás de mayor eficacia) si los vómitos interfieren con la administración oral de la NAC.
La restante terapéutica de soporte es la habitual en insuficiencia hepática grave e insuficiencia renal. Especialmente debe controlarse la hipoglicemia y los factores de coagulación. La vitamina K puede ser útil si hay hepatocitos viables.
El transplante hepático mejora el pronóstico en los casos con hepatitis fulminante. Los criterios para indicarlo se exponen en varias publicaciones, siendo la de Bernal42, la más reciente y la que agrupa criterios de varios grupos.
SALICILATOS
Constituyen un grupo de sustancias que se derivan del ácido salicílico y que se emplean desde hace más de cien años como analgésicos, antipiréticos y antiinflamatorios. El fármaco más importante de este grupo es el ácido acetilsalicílico, pero en nuestro país existen también otras sustancias comercializadas para uso oral o parenteral (acetilsalicilato de lisina, salicilamida, etc.), así como algunos preparados para uso tópico a base de ácido salicílico y salicilato de metilo. La descripción que se presenta a continuación hace referencia al ácido acetilsalicílico, pero los aspectos farmacocinéticos, clínicos y terapéuticos son también aplicables genéricamente al resto de los componentes de este grupo.
La intoxicación aguda por salicílicos era especialmente frecuente en pediatría en la década de los ochenta. Pero la progresiva substitución de la aspirina por el paracetamol y los envasados de mayor seguridad resistentes a la manipulación de los niños, han propiciado una menor disponibilidad y, por ello, una disminución de la frecuencia de intoxicaciones agudas por aspirina y derivados.
Cinética
El ácido acetilsalicílico es un ácido débil de bajo peso molecular (138 dalton). Se absorbe con rapidez en el estómago, en forma no disociada. En el intestino delgado la absorción es más lenta, aunque completa. La ingesta masiva de comprimidos puede inducir la formación de conglomerados que frenen su absorción. También hay un retraso en la absorción gástrica cuando se ingieren preparados comerciales de absorción intestinal así como en presencia de bicarbonato, ya que en ambos casos, la modificación del pH y el consiguiente aumento de las formas disociadas, disminuyen su capacidad de absorción. Otro factor de enlentecimiento de la absorción es el efecto inhibitorio que tiene la aspirina sobre el vaciado gástrico. También se absorbe por vía transdérmica.
A dosis terapéuticas, el pico plasmático se alcanza a las 1-2 horas pero en sobredosis no se logra hasta las 4-6 horas o incluso hasta las 12 horas o más.
La aspirina se hidroliza rápidamente a ácido salicílico a través de diversas esterasas: hepáticas, de la mucosa digestiva, del plasma , eritrocitarias, etc.43
A las dosis habituales, un alto porcentaje de aspirina circula unida a proteínas (80-90%) disminuyendo a un 70% o menos cuando se alcanzan concentraciones tóxicas que saturan los locus de fijación de la aspirina a la albúmina lo que da lugar a un rápido aumento de la fracción libre que es la activa.
El volumen de distribución es bajo, de 0,15 a 0,20 l/Kg alcanzando la mayoría de tejidos y líquidos orgánicos. La alcalosis al reducir la fracción salicílica no ionizada disminuye la capacidad de distribución. Lo contrario ocurre con la acidosis que aumenta la distribución y la toxicidad44.
La aspirina se hidroliza a nivel gastrointestinal a ácido salicílico. Su metabolización continúa a nivel hepático conjugándose con la glicina y el ácido glucorónico formándose diversos compuestos ácidos. Una vía secundaria es la oxidación que genera una serie de ácidos hidroxibenzoicos uno de los cuales es el ácido gentísico que a su vez se conjugará formándose finalmente ácido gentisúrico.
Una pequeña proporción (2,5%) de ácido salicílico no se metaboliza excretándose inalterado por vía renal. Los fenómenos de conjugación hepática son saturables por lo que a medida que aumenta la gravedad de la intoxicación se incrementará también la proporción de salicílico eliminado en forma libre a través de la orina. Todo ello influye en la semivida de eliminación, que a dosis terapéuticas es de 2-4 horas, mientras que en las intoxicaciones no tratadas puede alargarse a 18-36 horas.
La excreción renal se hace por filtración glomerular y secreción-reabsorción tubular distal que es altamente influenciable por cambios en el pH urinario45.
Mecanismo de acción
Sobre el SNC produce una estimulación directa que comporta alteraciones neurosensoriales, una estimulación del centro respiratorio que inducirá una hiperventilación con la correspondiente alcalosis respiratoria y la aparición de vómitos de origen central. En casos especialmente severos puede observarse una hipoventilación por depresión central en lugar de la hiperventilación.
Los efectos metabólicos son de gran trascendencia. Los salicilatos producen una inhibición de las deshidrogenasas del Ciclo de Krebs y un desacoplamiento de la fosforilación oxidativa mitocondrial que bloquea el paso de ADP a ATP y que tiene como consecuencia la disminución en la síntesis de ATP, el aumento del consumo de oxígeno y la disminución en la producción de anhídrido carbónico. Dosis más altas pueden dar lugar a una disminución del consumo de oxígeno con una menor capacidad de oxidacion celular . Ello provocará una mayor producción de ácido pirúvico y láctico y un incremento del metabolismo de ácidos grasos que generaran cuerpos cetónicos. El resultado final es la aparición de una acidosis metabólica y la producción de calor.
Debido a la mayor demanda metabólica se estimula la glucogenolisis hepática con aumento de la glucosa circulante. No obstante , si se agotan los depósitos de glucógeno hepático, aparecerá una hipoglicemia, especialmente en niños. Hay también una disparidad entre la glicemia plasmática y el descenso de la glucorraquia. Esta alteración podría explicar la aparición de convulsiones más frecuentes en los casos pediátricos46.
El equilibrio acido-básico presenta varias alteraciones. La alcalosis respiratoria pura debido a la hiperventilación es el trastorno más habitual y más precoz en las intoxicaciones leves o moderadas en el adulto. Esta alcalosis tiende a compensarse a través de la excreción renal de bicarbonatos, acompañándose de pérdidas de sodio, potasio y agua. Todo ello favorece la aparición de deshidratación, hipopotasemia y un menor efecto tampón del plasma.
En el curso evolutivo aparece una acidosis metabólica por acumulación de ácidos orgánicos y por los propios metabolitos del ácido acetilsalicílico. En pediatría es más frecuente observar esta acidosis inicialmente, sin la alcalosis respiratoria, más propia de las intoxicaciones de los adultos.
La aparición de una acidosis mixta por el desarrollo de una acidosis respiratoria (por depresión respiratoria o por edema pulmonar) es siempre un signo de gravedad. En el caso de vómitos repetidos, junto a los demás trastornos, aparecerá una alcalosis metabólica.
La hiperventilación, la hipertermia y los vómitos pueden producir, además, una grave deshidratación.
Las alteraciones sobre la hemostasia pueden generar una mayor fragilidad capilar, disminución de la agregación plaquetaria y descenso del tiempo de protrombina por disminución del factor VII. Todo ello se pone particularmente de manifiesto en pacientes con consumo crónico del fármaco más que en las intoxicaciones agudas.
Para tratar de contrarrestar el aumento de la producción de calor de origen metabólico se genera una vasodilatación y una hipersudoración, soliendo predominar el segundo efecto sobre el primero. En caso de hipertermia severa, a su vez, puede ser causa de rabdomiolisis47. A nivel gastrointestinal, la irritación de la mucosa puede causar naúseas, vómitos, hemorragia (poco frecuente), aparte de una acción específica sobre el vaciado gástrico en forma de espasmo pilórico y un menor peristaltismo. Las alteraciones de la glucosa y del metabolismo proteico sobre la endolinfa y la perilinfa pueden explicar los efectos otorrinolaringológicos (tinnitus e hipoacusia).
Manifestaciones clínicas
La edad del paciente (mayor riesgo en niños y ancianos) y, obviamente, la dosis ingerida son los factores que más influyen en el cuadro clínico.
Una sobredosis menor de 150 mg/Kg no debería provocar clínica, pero si la dosis aumenta a 150-300 mg/Kg las manifestaciones serían leves o moderadas, entre 300-500 mg/Kg serían ya graves o muy graves y potencialmente mortales al superar los 500 mg/Kg Así pues aparece una toxicidad significativa en el adulto tras una sobreingesta de 18-20 g de aspirina en una toma única.
El cálculo de la dosis ingerida no coincide necesariamente con la cantidad absorbida. En este sentido, se considera que más de 10 g en el adulto y más de 100 mg/ Kg en el niño son las cantidades mínimas que deben absorberse para provocar sintomatología, en general, de carácter leve.
Las manifestaciones clínicas más precoces incluyen manifestaciones digestivas (náuseas, vómitos), sensación de calor, rubefacción, sudoración, tinnitus con o sin hipoacusia. El tinnitus y la hipoacusia son, en general, los síntomas iniciales48 aunque su ausencia no excluye el diagnóstico ni predicen una mayor toxicidad posterior.
A medida que aumenta la concentración de salicílicos aparecen también, de forma relativamente precoz, otros síntomas y signos neurológicos: cefalea, vértigo, sordera, hiperventilación y , a veces, febrícula. Los casos más severos cursan con una encefalopatía caracterizada por hiperactividad, agitación, estado confusional, alucinaciones y lenguaje incoherente. A esta fase de confusión-agitación le sigue un estado de depresión de conciencia que varía desde una somnolencia o letargia hasta un coma. Pueden objetivarse hiperreflexia y convulsiones.
En general, la clínica confusional encefalopática, con o sin agitación pero manteniendo preservado el nivel de conciencia, es más característica de los adultos, sobre todo los ancianos. Pero en cambio, y con un grado de toxicidad similar, en los casos pediátricos aparecen coma y convulsiones con mayor rapidez y frecuencia49.
Junto a la toxicidad neurológica ya expuesta, la intoxicación aguda por salicílicos cursa con otras dos manifestaciones clínicas características: las alteraciones del equilibrio ácido-base y los trastornos metabólicos.
La alcalosis respiratoria es el desequilibrio ácido-base más precoz y más frecuente y que puede ser el único en los casos de toxicidad moderada. Como consecuencia de la alcalosis aumentará la excreción de sodio, potasio y bicarbonato (pH urinario > 6). Progresivamente aparecerá una acidosis metabólica que podrá compensar total o parcialmente la alcalosis.
Finalmente, las intoxicaciones muy severas cursan con una acidosis respiratoria o mixta importante que siempre es signo de mal pronóstico50.
El trastorno metabólico mas común es la hipertermia casi siempre moderada Pueden también observarse hipoglicemia (sobre todo en niños), hiperglicemia, hipoglicorraquia cetonuria e hipocalcemia. Además de lo antedicho, y de forma excepcional, puede presentarse un edema agudo de pulmón no cardiogénico, e insuficiencia renal (por deshidratación, rabdomiolisis o tubulopatía).
Diagnóstico
Al igual que otras intoxicaciones agudas que cursan, en general, sin deterioro de conciencia, la anamnesis y la clínica indican un diagnóstico de alta presunción en una elevada proporción de casos.
Las manifestaciones digestivas, el tinnitus y la hipoacusia con o sin hiperventilación, constituyen síntomas-guía que por su precocidad y relativa especificidad, son también útiles como apoyo diagnóstico.
Las exploraciones complementarias serán las que acabarán confirmando la intoxicación. Las de mayor utilidad son las siguientes:
– Reacción del cloruro férrico. Se añaden unas gotas de una solución al 10% de cloruro férrico a un cc de orina. La aparición de un color violáceo indica la presencia de salicílicos. El test tiene una alta especificidad (75,4%) y sensibilidad (93,8%). Un resultado positivo demuestra la presencia de salicílicos no necesariamente en sobredosis. Sólo hay dos posibles falsos positivos que corresponden a los ácidos acetoacético y fenilpirúvico51.
– Salicilemia, equilibrio ácido-base y electrolitos. Estas determinaciones analíticas no sólo son de ayuda diagnóstica sino que también tienen un interés pronóstico y terapéutico. La salicilemia confirma el diagnóstico aunque los niveles plasmáticos no se corresponden de una forma precisa con la toxicidad. Ello es debido a que cuando la intoxicación cursa con una acidemia importante, parte de los salicílicos abandonan el espacio vascular, desciende la salicilemia pero aumenta su distribución tisular y consecuentemente su toxicidad. La valoración de la salicilemia debe hacerse en base a una muestra obtenida al menos 6 horas post-ingesta, repetirla a las 4 ó 6 horas para comprobar su posible incremento, y evaluarla en función de la acidemia. A modo orientativo, una salicilemia (a las 6h de la ingesta) inferior a 500 mg/l probablemente indica una intoxicación leve, entre 600-900 mg/l es compatible con una clínica grave y si es mayor de 900 mg/l es potencialmente muy grave o incluso letal.
En cuanto a los trastornos ácido-base, la secuencia de mayor a menor gravedad son: acidosis mixta, acidosis metabólica y alcalosis respiratoria.
– Radiografía abdominal. La radiografía abdominal raramente puede ser de ayuda diagnóstica. Ocasionalmente pueden observarse o bien la radioopacidad de comprimidos de disolución entérica o bien, a través de un sorbo de papilla baritada, un bezoar de pastillas intragástrico.
En el diagnóstico diferencial, deberán tenerse en cuenta dos posibles errores. En primer lugar, la aparición de hipertermia, con una alteración del estado mental y una acidosis a pesar de ser compatible (especialmente en el anciano) con una intoxicación aguda o crónica por salicílicos puede etiquetarse erróneamente como patología infecciosa, retardándose el diagnóstico correcto.
De igual forma, un cuadro clínico que curse con hiperventilación, cetoacidosis, hiperglicemia y coma puede confundirse con una diabetes descompensada.
Tratamiento
Es excepcional que la intoxicación salicílica precise maniobras de reanimación en la atención inicial del paciente ya que las manifestaciones de máxima gravedad (convulsiones, edema pulmonar, acidemia, etc.) son muy poco frecuentes y aparecen, en general, en el curso evolutivo de la intoxicación.
Descontaminación digestiva
Cualquier método de descontaminación puede estar indicado al tener una eficacia similar si las condiciones clínicas son idénticas. No obstante, se prefiere el carbón activado al lavado y a la ipeca, pues ésta puede, teóricamente, propiciar la aparición de convulsiones.
En cuanto al aspirado-lavado gástrico puede utilizarse sin objeciones aunque debería reservarse para aquellos casos en que se sospechara la existencia de un conglomerado de comprimidos en la cavidad gástrica.
La administración de carbón activado debe basarse en los siguientes requisitos:
– Cantidad ingerida dentro del rango tóxico.
– Tiempo transcurrido desde la ingesta de 1-2 horas. Este intervalo puede prolongarse varias horas debido al retraso habitual en la absorción de la aspirina en sobredosis o en el caso de haber ingerido tabletas de cubierta entérica.
– La dosis de carbón activado debe ser lo mas próxima posible al ratio carbon/tóxico de 10:1.
– No hay datos concluyentes para avalar dosis repetidas de carbón activado53.
– No obstante, debe administrarse en general, una segunda dosis de carbón para evitar la desadsorcion53 y, siempre, en el caso de ingesta de comprimidos entéricos.
Aumento de la excreción. Alcalinización
La ionización de un ácido débil es pH dependiente. En el caso de la aspirina a un pH de 3, un 50% de las moléculas estarán ionizadas aumentando este porcentaje a medida que se incremente el pH.
Después de filtrarse a través del glomérulo, tanto las formas ionizadas como las no ionizadas se excretan por el túbulo renal, pero mientras las no ionizadas son reabsorbidas con facilidad no sucede así con las formas ionizadas. El resultado neto es que a mayor ionización menos reabsorción tubular y, por lo tanto, mayor excreción renal de salicílico.
Para lograr esta mayor ionización-excreción debe alcalinizarse la orina. Este efecto comenzará cuando la orina alcance un pH de 7,5 y será óptimo con un pH de 8-8,5.
Está demostrado54 que este aumento del aclaramiento renal (pasando de 1,3 ml/min a 100 ml/min) depende más de lograr un pH suficientemente alcalino y, en cambio, depende mucho menos de un incremento del filtrado glomerular a través de un mayor aporte hídrico. Por ello, para aumentar la excreción, no será necesario aplicar una diuresis forzada alcalina como era habitual hace un tiempo, sino que es suficiente con alcalinizar la orina a un pH óptimo. Esta alcalinización urinaria debe utilizarse sólo en los casos de toxicidad moderada o grave (salicilemia es superior a 500 mg/l en adultos y 350 mg/l en niños)55.
La importancia de la alcalinización no reside sólo en lograr un aumento de la excreción renal del tóxico, sino que, además, al corregir la acidemia cambiará el grado de ionización favoreciendo la permanencia del salicílico en el espacio vascular evitándose o disminuyendo su distribución en el SNC que, en definitiva, es lo que provoca la máxima gravedad.
La administración de bicarbonato sódico está indicado en casos de toxicidad significatica que habitualmente corresponden a salicilemias superiores a 500 mg/l.
Las intoxicaciones que cursan con alcalosis respiratoria pura con probable alcaluria y alcalemia no precisan alcalinización. En los pacientes con toxicidad moderada-grave con acidosis metabólica y alcalosis respiratoria, la alcalinización deberá, en todo caso, ser muy cautelosa. La pauta de administración dependerá de la severidad de las manifestaciones clínicas incluyendo el nivel de salicilemia y del grado de alteración del equilibrio ácido-base.
En caso de acidemia (pH inferior a 7,20) pueden administrarse un bolus de 1-2 mEq/Kg de bicarbonato sódico continuando con una perfusión a medida para lograr y mantener un pH urinario de 7,5. Una dosificación apropiada podría ser la siguiente:
– 100 ml de bicarbonato sódico 1M en perfusión continua en 4 horas.
– Simultáneamente, y, en otra vía intravenosa, 500 ml de glucosado al 5% más 20 mEq de ClK tambien en perfusión continua en 4 horas.
– Repetir este ciclo las veces necesarias añadiendo bolus horarios de 10-20 mEq de bicarbonato si el pH urinario cae por debajo de 7,5. Control riguroso de la potasemia.
– Suspender la alcalinización si el pH arterial supera 7,5.
La alcalinización debe ser muy juiciosa ya que comporta los siguientes riesgos: excesiva alkalemia, hipernatremia, hipokaliemia, hipocalcemia e hiperhidratación favorecedora de edema cerebral y edema pulmonar. Se precisan, por tanto, controles clínicos y analíticos rigurosos y frecuentes (por ejemplo cada 4 h.)
Obviamente, si existen ya signos compatibles con edema pulmonar o cerebral la alcalinización puede ser perjudicial siendo la hemodiálisis la técnica extractiva indicada.
La pauta alcalinizante puede suspenderse al observarse mejoría clínica, analítica y un descenso de la salicilemia por debajo de los 350-400 mg/l.
Aumento de la excreción. Hemodiálisis
La depuración extrarrenal debe ser considerada cuando concurren una serie de criterios clínicos y analíticos:
– Insuficiencia renal o insuficiencia cardíaca grave.
– Edema agudo de pulmón.
– Severa diselectrolitemia y / o acidosis refractarias al tratamiento.
– Signos persistentes de toxicidad sobre el SNC (convulsiones, confusión, disminución de la conciencia, edema cerebral).
– Deterioro clínico importante a pesar del tratamiento.
– Salicilemia superior a 1000 mg/l en caso de intoxicación aguda o superior a 600 mg/l en intoxicaciones crónicas.
A pesar de que con la hemoperfusión se puede lograr un aclaramiento renal de más de 80 ml/min56 superior al obtenido por la hemodiálisis, esta técnica es la de elección ya que además de alcanzarse una buena excreción, corrige las alteraciones hidroelectrolíticas y los trastornos ácido-base.
Tratamiento inespecífico
Fluidoterapia. Los pacientes con intoxicación moderada-grave habitualmente presentan diversos grados de deshidratación. Debe administrarse suero glucohiposalino (10-20 ml/Kg/1-2 h) con los necesarios controles hasta obtener una buena respuesta diurética.
Diselectrolitemia. Deben seguirse controles periódicos. La alteración más significativa es la hipopotasemia que puede estar enmascarada por la acidosis o acrecentada al alcalinizar.
Hipocalcemia. Puede observarse un descenso del calcio iónico en casos graves acentuándose con la alcalinización. Se precisa control periódico, monitorización cardíaca y sales de calcio si es necesario.
Hipertermia. Usar enfriamiento externo si la temperatura rectal es superior a 40º.
Glucosa. Administrar 50 ml de glucosa hipertónica (33-50%) siempre que se produzca una disminución de la consciencia, una encefalopatía o convulsiones a pesar de comprobarse una glicemia normal ya que puede existir una hipoglucorraquia.
Convulsiones. Aparte de la glucosa, administrar una benzodiacepina con cautela ya que la sedación puede modificar la alcalosis respiratoria agravando el cuadro.
Enema pulmonar. Al ser no cardiogénico no es tributario de diuréticos y digoxina. Debe administrarse oxígeno, intubación y PEEP.
Enema cerebral. Aplicar tratamiento convencional
Coagulopatía. Los posibles trastornos del tiempo de protrombina o del INR, en general, son asintomáticos. Puede administrarse vitamina K. En caso de sangrado activo, plasma fresco.
ÁCIDO VALPROICO
El ácido valproico (AV) y sus derivados (fundamentalmente el valproato sódico) han sido incluidos a partir de 1978 en el arsenal terapéutico anticomicial, inicialmente como monoterapia en el tratamiento de las ausencias y junto a otros anticonvulsivantes en el manejo de otros tipos de crisis más complejas bien parciales o generalizadas57. Con posterioridad han ido incrementándose sus indicaciones a cierto tipo de desórdenes afectivos58, como profilaxis y terapia de fase aguda de la migraña59, para el control del dolor neuropático60 y a partir de 1995 y motivado en gran medida por el ajustado rango terapéutico del litio, la FDA ha autorizado su empleo en la manía asociada a trastorno bipolar61.
Conforme ha ido ampliándose el capítulo de indicaciones lo ha ido haciendo igualmente el de intoxicaciones, habiéndose cuantificado en EE.UU. un incremento del 707% para el decenio 1990-200062, con un total de 5.204 recogidos, 3.880 de los cuales obedecen a intentos autolíticos, 373 a casos de mayores tasas de toxicidad y 16 a muertes63.
Farmacocinética y farmacodinamia
Tras la dosis terapéutica el AV es rápidamente absorbido por el tracto gastrointestinal, con un pico sérico alcanzable tras 1-4 horas post-ingestión del comprimido64, 15 a 60 minutos tras el jarabe y de 3 a 7,5 horas tras la toma del comprimido entérico retard65.
Aunque controvertido, el rango terapéutico se encuentra comprendido entre 50 y 100 µg/ml66. El 90% está unido a proteínas plasmáticas, aunque esta relación es dependiente de la concentración sérica, siendo del 81,5% para niveles de 130 mg/ml y de tan solo el 35% cuando se superan los 300 µg/ml67. El incremento de la fracción libre conlleva mayor distribución del mismo a los órganos diana, aumentando los efectos clínicos para una misma concentración sérica.
La vida media oscila entre las 7 y 15 horas68, incrementándose la misma en ancianos, cirróticos y neonatos y disminuyendo cuando se asocia a otros fármacos anticomicales, especialmente los inductores de enzimas (fenobarbital, fenitoína, carbamacepina). Es metabolizado predominantemente en el hígado, siendo excretado menos del 3% por la orina sin modificar69. A nivel hepático destaca la glucuronización directa y la excreción biliar, con posible recirculación enterohepática70. La eliminación sigue una cinética de primer orden, con aclaración plasmática tras dosis terapéutica de 5 a 10 ml/min, independientemente del flujo sanguíneo hepático, siendo el aclaración de droga libre de 77 ml/min71.
Mecanismo de acción
La propiedad anticonvulsivante parece estar relacionada, al menos en parte, por el incremento regional de concentraciones del ácido gamma-aminobutírico (GABA), el principal inhibidor de la transmisión cerebral72, presumiblemente por bloqueo de la GABA transferasa y succinil aldehído deshidrogenasa. Igualmente puede prolongar el tiempo de recuperación de los canales inactivados de sodio y tener efecto en los canales de potasio presentes en las membranas neuronales, aunque no parecen contribuir significativamente en sus efectos anticomiciales73.
Toxicidad
No son infrecuentes los efectos tóxicos asociados a dosis terapéuticas, en especial cuando la cantidad diaria ingerida se aproxima a 1800 mg, con niveles sanguíneos asociados superando los 100 µg/ml, aunque la correlación entre concentración plasmática y efectos clínicos no está suficientemente definida74.
Intoxicación aguda
La mayoría de casos documentados hacen referencia a la sobredosificación de AV por vía oral. En estas situaciones, y con independencia de la formulación ingerida, se han descrito picos tardíos en plasma de hasta 18 horas con una media de 7,4±3,9 h75. Ingels y col76 apreciaron cómo en el 13% de los pacientes, que posteriormente presentarían niveles plasmáticos tóxicos > 120 µg/ml, sus niveles de AV al ingreso eran bajos o indetectables.
Es por ello justificada la recomendación de determinaciones seriadas de AV en situaciones de sobredosis, planteando el alta tras constatar la tendencia descendente de los mismos una vez alcanzado el rango terapéutico. En cuanto a la presentación clínica, la intoxicación medicamentosa por AV comparte gran similitud genérica con la asociada a otros anticomiciales, aunque con características específicas propias. Domina la clínica secundaria a la afectación del SNC, presenta alteraciones metabólicas específicas y el resto de sintomatología (hepática, pancreática y hematológica) no guarda relación con los elevados niveles plasmáticos.
La depresión del SNC constituye la alteración clínica fundamental en la intoxicación por AV, siendo frecuente la presencia de coma de alto grado que requiere instaurar intubación y ventilación mecánica77. Estudios multicéntricos78 refieren porcentajes del 71% de letárgicos al ingreso, mientras el 15% lo hacían en coma. Además, el 100% de los pacientes con niveles plasmáticos superiores a 850 µg/ml presentaban coma, requiriendo intubación el 63%. Tras sobredosificación, el coma se ha comprobado que persiste a los días de haber normalizado los niveles plasmáticos, lo que induce a pensar que son los metabolitos del AV los responsables de perpetuar la situación depresora del SNC79.
Edema cerebral se ha observado tanto en sobredosis como en terapias crónicas mantenidas con dosis supraterapeúticas80. Se describe a las 48-72 horas de la ingestión81 y su mecanismo de formación es controvertido, implicando a los metabolitos en la génesis del mismo82, la hiperamoniemia coincidente80 o la estimulación de los receptores NMDA83.
En cuanto a las manifestaciones tóxicas cardiovasculares, puede aparecer inestabilidad hemodinámica (hipotensión) en los casos de ingestión masiva. El 25% de los pacientes con niveles séricos superiores a 850 µg/ml desarrollaron episodios de hipotensión, con taquicardia asociada en el 17%78. A nivel digestivo, se han visto pancreatitis asociadas tanto a la ingesta prolongada como a episodios de sobredosis84. Episodios de hepatotoxicidad son menos frecuentes en situaciones agudas. En el caso de consumo crónico sí que se han detectado elevaciones de transaminasas sin clínica asociada en hasta el 44% de los pacientes85 y episodios idiosincráticos de hepatitis rápidamente progresiva con hallazgos histológicos similares a los detectados en el síndrome de Reye86.
Metabólicamente puede aparecer hipernatremia y acidosis metabólica con anión gap elevado, a partir de valores plasmáticos superiores a 450 µg/ml87. Ha sido descrita la presencia de acidosis láctica e hipocalcemia marcada67, aunque la primera precisa descartar el compromiso hemodinámico como desencadenante primario del evento y la segunda no excede en su ratio del 6% de los casos88, la mayoría de los cuales con niveles plasmáticos superiores a 450 µg/ml. Situaciones de fracaso renal agudo han sido cuantificadas en el 1% de los casos78.
La trombocitopenia es la manifestación de toxicidad hematológica más frecuente, seguida por la leucopenia y en menor grado por la anemia. La incidencia constatada de las mismas es del 8% en la primera y del 3% en la segunda78.
Tratamiento
Como en cualquier otra situación de intoxicación, máxime si se acompaña de depresión asociada del nivel de conciencia, es mandatorio el control de la vía respiratoria con intubación precoz y oxigenoterapia. Análogamente el acceso venoso, estabilización hemodinámica y monitorización de funciones vitales es común a todas ellas.
Las determinaciones analíticas obligadas incluyen la cuantificación plasmática de AV y la valoración del estado de oxigenación tisular. Recordar que dado el pico tardío presente en los casos de intoxicación aguda es necesario realizar perfil evolutivo de los niveles plasmáticos. Asociar determinaciones de urea, creatinina, glucosa, calcio, osmolaridad sérica, amonio, transaminasas, bilirrubina, amilasa, lipasa, hemograma y coagulación.
A todo paciente con sospecha de intoxicación por AV se debe realizar lavado gástrico y administración de carbón activado dado el retraso en alcanzar el pico plasmático en ingestión significativa, la posible presencia de presentaciones farmacéuticas de liberación retardada y la posibilidad de circulación enterohepática.
El alto grado de unión a proteínas condiciona la escasa eficacia de eliminación extracorpórea a concentraciones terapéuticas. Rebasado el umbral plasmático de los 300 µg/ml es cuando modalidades de hemodiálisis y/o hemoperfusión son consideradas opciones viables88, habiéndose planteado últimamente la técnica de hemodiálisis de alto flujo como monoterapia89. El valor sérico absoluto o el grado de coma no debería ser utilizado como determinante para el empleo de técnicas de depuración extrarrenal, valorándose igualmente la presencia de inestabilidad hemodinámica con persistente acidosis metabólica no reversible con fluidoterapia como factores primarios a resolver mediante la homeostasis lograda a partir de técnicas dialíticas.
La naloxona (0,01 mg/Kg) ha demostrado su utilidad en intoxicaciones por AV que presentaban características clínicas similares a la intoxicación por opiáceos (miosis, coma, depresión respiratoria) y que fue lo que inicialmente motivó su utilización, confirmándose a posteriori la inexistencia de los segundos y la presencia del primero90. Posteriormente se ha comprobado la dudosa utilidad del fármaco en intoxicaciones con niveles séricos de AV superiores a 190 µg/ml91.
La l-carnitina, a partir del hallazgo de su depleción en pacientes tratados crónicamente con AV, ha sido incluida como suplemento por vía oral ( hasta un máximo de 2 g/día ). En situaciones de intoxicación o hiperamonemia, la administración intravenosa de carnitina a razón de 150-500 mg/Kg/día (hasta 3 g/día) ha sido recomendada92. Con independencia de la frecuencia en su empleo, dada la buena tolerancia y los potenciales beneficios antiedema cerebral secundario a la hiperamonemia parece razonable su utilización en situaciones de intoxicación por AV.
BARBITÚRICOS (FENOBARBITAL)
Los barbitúricos constituyeron una causa frecuente de intoxicación hasta la década de los años 60. A partir de entonces su uso ha ido decreciendo y han sido sustituidos por drogas menos tóxicas. A pesar de ello siguen viéndose casos de suicidio, muerte accidental y adicción, mayormente por uso ilícito93. Las principal intoxicaciones suelen estar en relación con la toma de pentobarbital, secobarbital, amobarbital, siendo menos frecuentes los casos de toxicidad debidos a tiopental, barbital y pentobarbital.
Los barbitúricos se clasifican según su duración de acción en :
– Acción prolongada: fenobarbital, barbital.
– Acción intermedia y corta: pentobarbital, secobarbital.
– Acción ultracorta: tiopental.
Farmacocinética
Su absorción a través del tracto gastrointestinal se realiza por difusión pasiva no iónica, su velocidad de absorción depende del pH gástrico e intestinal, del pK del barbitúrico y del grado de solubilidad. A partir de los 30 minutos pueden detectarse niveles en sangre aunque el pico se alcanza a las 4 horas. Por la gran liposolubilidad de su forma no ionizada atraviesan bien las membranas biológicas. La mayoría son metabolizados en el hígado, induciendo al sistema microsomal P450 que afecta a su propio metabolismo y al de otros fármacos. Los más liposobles siguen un proceso de glucoronización antes de ser excretados por el riñón. Los de más larga acción son menos metalizados por el hígado94. El fenobarbital con un pK de 7,2 es fundamentalmente excretado por el riñón, incrementándose su eliminación con la alcalinización urinaria. Globalmente siguen una cinética de primer orden a bajas concentraciones y una cinética de orden cero a altas concentraciones95.
La fijación a proteínas plasmáticas es baja, siendo para los barbitúricos de acción prolongada o intermedia del 5-20%, y para los de acción corta y ultracorta superior al 35%.
El volumen de distribución oscila desde 0,6 l/Kg para el fenobarbital hasta 2,6 l/Kg para el tiopental.
Las dosis tóxicas oscilan entre 3-6 gramos para los barbitúricos de corta acción y entre 6-10 gramos para los de larga acción96. El nivel anticonvulsivante para el fenobarbital está entre 10-40 µg/ml y en caso de que se empleen dosis de carga intravenosa (15-25 mg/Kg) hay que tener presente que la formulación contiene un 60% de propilenglicol, por lo que no se debe superar la velocidad de infusión de 60 mg/min, además a las dosis antes indicadas es preciso la intubación endotraqueal.
Acción farmacológica
La combinación de urea y ácido malónico dan lugar al ácido barbitúrico, molécula que carece de actividad hipnótica y a partir de la cual se han ido sintetizando los diferentes tipos de este compuesto. La potencia de cada droga es función de la constante de ionización y del grado de liposolubilidad. A mayor liposolubilidad mayor potencia y a pH plasmático más bajo mayor entrada de la forma ionizada en el cerebro. Ocasionan depresión global del SNC, ya que aumenta la actividad inhibidora del GABA, suprimiendo la transmisión neuronal . Actúan sobre la formación reticular del tronco cerebral con disminución del nivel de conciencia generalizada aunque a veces con focalidad, hasta llegar al coma97. Todos los barbitúricos son similares farmacológicamente y difieren sólo en la velocidad de comienzo y duración de su acción. El alcohol tiene un efecto aditivo en la potenciación de los efectos de los barbitúricos.
Produce un efecto depresor sobre los centros respiratorios, suprimiéndose la respuesta hipóxica con dosis menores que el impulso quimiorreceptor del CO2. Ocasionan disminución de las resistencias vasculares sistémicas y del gasto cardíaco con hipotensión severa.
A nivel intestinal se produce una disminución del tono y peristaltismo, con íleo y disminución de la absorción. Hasta en un 6% de los pacientes se desarrolla una epidermólisis bullosa en zonas de pliegues y de presión (también puede acontecer en intoxicaciones por antidepresivos tricíclicos, metadona y monóxido de carbono)98.
Clínica
Se considera que el paciente está gravemente enfermo cuando presenta uno de las siguientes situaciones: coma; depresión respiratoria; fallo hemodinámico; trastornos cutáneos (livideces, escarificación, eritemas, bullas) e hipotermia.
El coma es proporcional a la dosis ingerida y se potencia por el alcohol99. El coma suele ser hipotónico, con ausencia de reflejos osteotendinosos y reflejo fotomotor. La depresión respiratoria en forma de apnea o respiración irregular es de presentación precoz.
La hipotermia por afectación del centro termorregulador es de mal pronóstico. A nivel hemodinámico hay hipotensión, taquicardia y mala perfusión periférica.
Las complicaciones respiratorias incluyen atelectasias, aspiración y neumonía. Puede haber rabdomiolisis e insuficiencia renal aguda.
Tratamiento
Los barbitúricos no dañan directamente el SNC, a no ser que coexistan situaciones de hipoxia o shock, por lo tanto existe un potencial de total recuperación con una adecuada terapia de soporte.
Los pacientes en situación de depresión respiratoria, cianosis o shock deben ser inmediatamente intubados y conectados a ventilación mecánica. En el resto de los pacientes si se va a realizar un lavado gástrico, requerirán aislamiento de la vía aérea cuando el reflejo de la tos está ausente o si hay dudas acerca de la adecuación de su mecánica respiratoria. Hay que monitorizar los gases arteriales para mantener una pO2> 80 mmHg (sO2> 94%).
La hipotensión es desencadenada por un volumen intravascular disminuido, hipoxia, acidosis y depresión de la función miocárdica. Debemos corregir el componente hipóxico y proceder a una expansión de volumen, con coloides a 20 cc/min intentando alcanzar presiones venosas centrales de 6 cm de H2O. Si no hay respuesta a la fluidoterapia se emplearán drogas vasoactivas para alcanzar presiones sistólicas superiores a 90 mmHg, vigilando si con ese nivel tensional la respuesta diurética es apropiada.
En cuanto al lavado gástrico, la retirada de la droga no absorbida es útil si no han pasado más de tres horas desde la ingestión. La única excepción son aquellos pacientes que han desarrollado un íleo como consecuencia de la cantidad ingerida de barbitúrico. El lavado se realizará previa intubación100. La inducción del vómito con jarabe de ipecacuana está contraindicada101.
Una vez evacuado el estómago y si hay ruidos intestinales administraremos carbón activado y un catártico: carbón activado 1 mg/Kg seguido por 25-50 g/4-6 horas durante 3-4 días hasta la desaparición de los síntomas102, y sorbitol, 50 gramos con 200 cc de agua, o alguna solución comercial estándar.
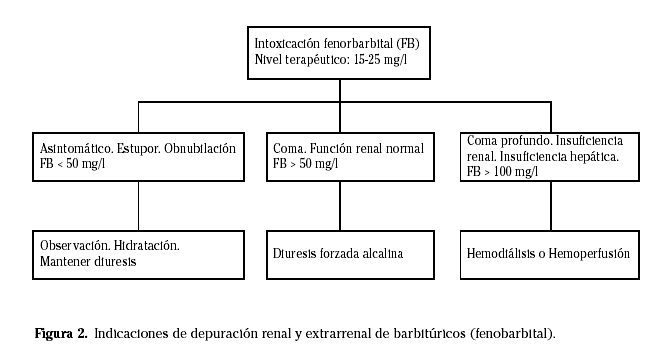
La diuresis forzada y en el caso del fenobarbital la alcalinización de la orina acelera la eliminación del barbitúrico. La hemodiálisis es más efectiva en la eliminación de los barbitúricos de acción larga e intermedia. Sus indicaciones son: fallo renal o hepático; shock o coma prolongado refractario al tratamiento; ingestión de dosis letales y un nivel sérico alto predictivo de coma prolongado103 (Fig. 2).
CARBAMAZEPINA
La carbamazepina (CMZ) es un agente psicotropo ampliamente utilizado como anticomicial en adultos y niños así como en el tratamiento de neuralgias, síndromes dolorosos crónicos, demencias y trastornos afectivos104, 105.
Farmacocinética y farmacodinamia
Administrada por vía oral, la CMZ es absorbida prácticamente en su totalidad de forma lenta y en ocasiones errática. Alcanzando el pico plasmático entre las 4 y 12 horas posteriores a la ingesta, llega a prolongarse de 24 a 72 horas tras sobredosis masivas106.
A concentraciones plasmáticas terapéuticas, 4 a 10 µg/ml, el 75% permanece unido a proteínas con un volumen de distribución de 0,8 a 1,4 l/Kg que aumenta a 0,96-2,07 l/Kg tras su empleo continuado107.
Es metabolizada en el hígado fundamentalmente por oxidación a partir de enzimas microsomales, destacando entre la variedad de sus metabolitos el CBZ 10,11 epóxido (CBZE) que constituye aproximadamente el 40% de los mismos, participando de las mismas propiedades anticomiciales y tóxicas que su predecesor, con menor afinidad a proteínas y menor volumen de distribución108.
Tras varias semanas de uso continuado la CMZ induce su propio metabolismo a partir de enzimas hepáticos P-450, CYP 3A3 y 3A4, lo que contribuye a modificar su vida media de 20-65 horas que reflejaría el inicio de la terapia en un paciente hasta entonces no consumidor de la misma, a las 8-17 horas presentes en el tomador crónico. La vida media del CBZE es de 8 a 14 horas109.
Terapias anticonvulsivantes asociadas del tipo fenóbarbital, fenitoína y ácido valproico pueden acelerar el metabolismo por inducción enzimática. Análogamente eritromicina, isoniacida, bloqueantes de los canales del calcio, cimetidina, ketoconazol y otros inhibidores de los CYP 3A3 y 3A4 pueden llegar a generar toxicidad con dosis terapéuticas.
Mecanismo de acción
Es superponible al de la fenitoína, reduciendo la permeabilidad de las membranas neuronales a los iones sodio y potasio. Inhibe la liberación del glutamato y otros neurotransmisores sinápticos y bloquea la recaptación de norepinefrina105, 110.
La CBZ es también un potente inhibidor de los receptores muscarínicos y nicotínicos, de los receptores adenosínicos cerebrales y por su similitud estructural con los antidepresivos tricíclicos interfiere en los canales del sodio miocárdicos111-113.
Toxicidad
Aparecen generalmente al sobrepasar los niveles plasmáticos de 12 µg/ml, destacando los referidos al SNC114, gastrointestinal115, cardiovascular116 y renal117. Entre los primeros, frecuentes al inicio del tratamiento, cabe destacar la presencia de vértigos, mareos, somnolencia, fotofobia, inestabilidad, ataxia, nistagmus, naúseas y vómitos.
Se han descrito descompensaciones de cardiopatía isquémica estable previa, fallo cardiaco congestivo, descompensación tensional, arritmias, bloqueos cardíacos, hiponatremias e intoxicaciones por agua.
Intoxicación aguda
Es importante hacer la diferenciación previa entre sobredosis en pacientes ya consumidores habituales del medicamento o en aquellos que lo ingieren por primera vez. Entre los segundos es esperable toxicidad significativa a partir de 20 a 30 mg/Kg, con situaciones de riesgo vital tras la ingestión de 140 mg/Kg. Por el contrario, entre consumidores crónicos, fruto del metabolismo inducido, han llegado a ser publicadas supervivencias tras la ingesta de 640 mg/Kg118, 119.
En situaciones de sospecha fundada de intoxicación por CBZ, y tras anamnesis, es importante valorar modificaciones recientes de la dosificación habitual en tomadores crónicos o su interacción con medicamentos que inhiban su metabolismo.
La exploración física deberá incidir, con independencia de la correcta valoración de signos vitales, en el tamaño, reactividad y posición pupilar, nivel de conciencia, respuesta a estímulos, reflejos, función cerebelar y peristaltismo intestinal, ECG, monitorización cardíaca, electrolitos y glucosa.
Resulta mandatoria la determinación de los niveles plasmáticos de CBZ, no tanto en su valor aislado como en la evolución cronológica de los mismos, obtención del pico sanguíneo y seguimiento hasta que los mismos desciendan a valores inferiores a 10 µg/ml120. El enlentecimiento en la absorción digestiva en situaciones de sobredosificación por formación de bezoar y desarrollo de íleo intestinal secundario al potente efecto anticolinérgico de la CBZ obligan a ello. De ahí la utilidad de realizar determinaciones asociadas del metabolito CBZE, estableciendo la relación CBZ/CBZE y considerando que cuando ésta supera el valor de 2,5 nos encontramos en permanente absorción del producto activo desde el aparato digestivo121, 122.
Diferentes situaciones clínicas se observarán en relación a la cantidad de producto activo ingerido destacando en las intoxicaciones severas la presencia de coma, convulsiones, hipotensión y alteraciones de la conducción cardíaca. En intoxicaciones moderadas domina la depresión del nivel de conciencia sin llegar a precisar medidas de soporte de vía aérea y en intoxicaciones menores podremos observar somnolencia, nistagmus, ataxia y disartria123.
Manifestaciones tóxicas neurológicas
La depresión del nivel de conciencia representa el hallazgo más frecuente en situaciones de intoxicación, llegando a alcanzar el 100% de los casos124. Fluctuaciones del mismo, con súbitas mejorías y empeoramientos125, son reflejo del errático patrón de absorción digestiva. La CBZ es un importante anticomicial a dosis terapéuticas pero constituye un potente proconvulsivante en sobredosis, de ahí su observación junto a otros hallazgos clínicos neurológicos como midriasis, anomalías del tono muscular y reflejos, ataxia, movimientos coreoatetósicos, hemibalismo, nistagmus y oftalmoplejia126.
Manifestaciones tóxicas cardiovasculares
Inicialmente se pesenta discreta taquicardia, hipertensión arterial seguida de hipotensión y anormalidades menores electrocardiográficas del tipo prolongación del espacio PR, QRS y QT127. Son frecuentes en pacientes de edad avanzada la pérdida de onda P, presencia de complejos ventriculares prematuros y bloqueos auriculoventriculares de diferente grado128.
Manifestaciones tóxicas digestivas y renales
Naúseas, vómitos, íleo intestinal y retención urinaria son signos de intoxicación. Se han constatado reacciones pancreáticas y disfunciones hepáticas transitorias129. Destacar la presencia de hiponatremia secundaria a síndrome de inadecuada secreción de ADH130.
Tratamiento
Las intoxicaciones moderadas plantean el diagnóstico diferencial con hipoglucemias, patología neurológica primaria, síndrome serotoninérgico e intoxicación por etanol. En las agudas cabe descartar intoxicación por antidepresivos tricíclicos, agentes anticolinérgicos, trauma y otros anticomiciales.
El tratamiento de la intoxicación por CBZ es esencialmente de soporte y tratamiento sintomático, precisando en casos severos de intubación orotraqueal y apoyo ventilatorio mecánico. La mayoría de fallecimientos están asociados a aspiración pulmonar y depresión respiratoria131.
En los casos de hipotensión arterial el tratamiento deberá incluir venoclisis con salino isotónico y en su caso vasopresores, prestando especial atención a la intoxicación acuosa frecuentemente asociada al efecto antidiurético de la CMZ. Las arritmias cardíacas conllevan terapia específica y las convulsiones responden a benzodiacepinas y fenobarbital, con dudosa respuesta a fenitoína132.
El empleo del carbón activado es el método recomendado de descontaminación digestiva en todo paciente sintomático. Se aconsejan múltiples dosis del mismo en situaciones de intoxicación masiva, preferiblemente con niveles plasmáticos superiores a 20 µg/ml133.
La diuresis forzada y los procedimientos hemodialíticos carecen de indicación. La hemoperfusión con carbón, aunque se ha constatado que aclara CMZ del torrente sanguíneo, dada su escasa proporción y las frecuentes dificultades técnicas asociadas, ha quedado relegada a situaciones extremas de elevados niveles plasmáticos, coma persistente y patente inestabilidad hemodinámica134.
FENITOÍNA
La fenitoína pertenece a la familia de las hidantoínas (a la cual también pertenecen la mefenitoína y la etotoína); fue sintetizada en 1908 por Bilz, e introducida como medicación anticonvulsivante en 1938, siendo desde entonces la medicación anticonvulsivante más frecuentemente usada135. Es efectiva para el tratamiento de crisis convulsivas tónico-clónicas generalizadas, parciales complejas y convulsiones focales. Se ha utilizado también en el tratamiento de la neuralgia del trigémino, de las convulsiones tóxicas y como profilaxis de convulsiones en el trauma craneal. Debido a sus propiedades antiarrítimicas (tipo IB) fue el antiarrítmico de elección de la intoxicación digitálica136, no siendo considerado de primera elección tras la aparición de los fragmentos Fab137.
Farmacología
La fenitoína actúa sobre las bombas y canales del sodio en todas las membranas excitables. A nivel neuronal inactiva los canales del sodio, siendo su efecto inhibitorio dependiente del voltaje y la frecuencia de descarga neuronal, suprimiendo la actividad neuronal repetitiva y previniendo su transmisión sináptica a neuronas adyacentes.
A nivel miocárdico tiene un efecto similar al de la lidocaína, inhibiendo los canales del sodio, disminuyendo el período refractario efectivo y la automaticidad de las fibras de Purkinge, con poco efecto sobre el QRS y la duración del potencial de acción.
Cinética
La absorción tras ingesta oral es lenta y variable especialmente a dosis tóxicas, por ello es necesario determinar niveles plasmáticos seriados tras sobredosis. Una vez absorbida se distribuye por todo el organismo con un volumen de distribución (VD) de 0,6 l/Kg. A nivel cerebral se obtienen concentraciones similares a las plasmáticas a los 10 minutos de inicio de una infusión intravenosa (siendo mayores en tronco cerebral y cerebelo que en el córtex cerebral)138. En el tejido miocárdico y en el líquido cefalorraquídeo se produce el equilibrio respecto a las concentraciones plasmáticas hacia los 60 minutos. Los picos séricos aparecen entre 2,6-8,9 horas tras la toma de un compuesto de liberación retardada (Dilantin®), pero la misma formulación ingerida en sobredosis puede tardar en alcanzar picos séricos hasta 12 días139.
Se une en un 90% a proteínas plasmáticas, principalmente a la albúmina, existiendo una fracción libre con la misma actividad biológica y tóxica que supone el 10% de los niveles plasmáticos. Está fracción libre de fenitoína esta incrementada así como su VD en todos aquellas situaciones donde sustratos endógenos o exógenos compitan con la unión a proteínas plasmáticas así como en casos de hipoalbuminemia. Estos pacientes pueden manifestar toxicidad a pesar de estar los niveles de fenitoína en rango terapeútico, siendo necesario por tanto la medición de los niveles de fracción libre. En los pacientes hipoproteinémicos podemos corregir los niveles de fenitoína sabiendo el nivel de albúmina. Para calcular la concentración de fármaco (C normal) que estaría presente en un paciente con nivel de albúmina normal, se puede aplicar la siguiente ecuación:

Metabolismo
Es fundamentalmente hepático. Un 70% es transformada a un metabolito derivado del parahidroxifenilo, siendo posteriormente conjugada con un glucorónido que la hace más soluble en agua, secretada en la bilis, reabsorbida y posteriormente excretada en la orina (sólo un 4% es excretada en orina de forma inalterada)140. El sistema enzimático de la parahidroxilación es saturable. A concentraciones inferiores a 10 µg /ml sigue una cinética de primer orden (un porcentaje fijo de la droga se metaboliza por unidad de tiempo), con una vida media de 22 horas. Por encima de 10 µg/ml la metabolización se satura y sigue una cinética de orden cero (se metaboliza una cantidad fija por unidad de tiempo) prolongándose su vida media. A su vez el aumento o inhibición enzimática del metabolismo de la fenitoína por causas genéticas, enfermedades hepáticas o tratamiento concomitante con otras drogas pueden incrementar o disminuir sus niveles séricos.
Niveles plasmáticos y toxicidad
Se pueden presentar signos de toxicidad aguda con una única toma igual o superior a 20 mg/Kg. Los niveles plasmáticos terapéuticos de fenitoína son de 10-20 µg /ml y el de la fracción libre de 1-2 µg/ml. Los pacientes con enfermedad cerebral previa son más propensos a la toxicidad con niveles bajos. En general existe una buena correlación entre los niveles plasmáticos y la toxicidad, tanto en sobredosis agudas como crónicas141, ya que no se ha descrito tolerancia a la fenitoína (Tabla 3).

Casi todos los pacientes con convulsiones inducidas por fenitoína tienen niveles plasmáticos mayores de 30 µg/ml. Pueden aparecer convulsiones paradójicas, en general en pacientes epilépticos y que en ocasiones no se acompañan de otros signos de toxicidad. Se presentan cuando aumentan los niveles del fármaco y desaparecen cuando disminuyen142.
Clínica
El comienzo de los signos y síntomas ocurre en horas tras la ingestión de una sobredosis. El sistema nervioso central es el principal afectado143, objetivándose los efectos de la inhibición cortical y de la excitación cerebelo-vestibular. Las manifestaciones más precoces de toxicidad siguiendo a una sobredosis son nistagmo horizontal en la mirada forzada (su ausencia no excluye una severa intoxicación), ataxia y adormecimiento. Con intoxicaciones más severas, nistagmo vertical o bidireccional, disartria, progresiva ataxia hasta el punto de no poder andar, padecer hiperreflexia y disminución del nivel de conciencia. En grandes sobredosis puede haber coma y/o depresión respiratoria (es raramente observado y deberemos considerar a su vez otras alternativas diagnósticas144 (Tabla 4).
A nivel cardiovascular no se ha descrito ningún caso de toxicidad cardíaca en paciente con sobredosis vía oral. La administración intravenosa produce hipotensión con disminución de las resistencias vasculares sistémicas, depresión de la conducción cardíaca que puede progresar al bloqueo cardíaco, taquicardia y fibrilación ventricular. Es más frecuente la aparición de este tipo de complicaciones en gente mayor con cardiopatía de base145.
Tratamiento
La intoxicación raramente es mortal. Es preciso iniciar los cuidados de soporte general, monitorización de constantes vitales y descontinuar la fenitoína. No existe antídoto.
Si hay disminución de conciencia se administrará glucosado al 50%, tiamina y naloxona (evitar el uso de flumazenilo si hay antecendentes de convulsiones). En caso de convulsiones tratamiento con benzodiacepinas.
El lavado gástrico se hará como siempre tras protección de vía aérea en pacientes con bajo nivel de conciencia, riesgo de broncoaspiración y/o convulsiones. Administrar múltiples dosis de carbón activado (circulación enterohepática de la fenitoína) 1g/Kg de peso cada 24 horas.
La diuresis forzada, hemodiálisis y hemoperfusión son inefectivas146.
Evolución y pronóstico
Normalmente el paciente evoluciona con una progresiva resolución de los signos y síntomas de la intoxicación, llegando a una completa resolución. La muerte es rara después de la sobredosis de fenitoína. Se ha descrito en casos en relación con su administración intravenosa para el tratamiento de arritmias cardíacas en ancianos y aisladamente niños tras sobredosis oral que ocasionó coma e hipotensión.
ISONIACIDA
La isoniacida (INH) es el fármaco de elección para el tratamiento de la tuberculosis y para profilaxis en pacientes con test de la tuberculina positivo. Prevalece su uso en pacientes con síndrome de inmunodeficiencia adquirida, en adictos a drogas por vía parenteral, en alcohólicos, en el sudeste asiático, mejicanos e indios americanos.
Cinética
La droga es normalmente administrada por vía oral siendo casi complemente absorbida (90-95%). Las concentraciones pico se alcanzan entre 1-2 horas postingestión, estimándose que el pico plasmático de concentración tras una dosis de 5 mg/Kg es de 5 mg/l.
El volumen aparente de distribución es de 0,6 l/Kg. Su unión a proteínas plasmáticas es del 10-15%147. Se distribuye por todos los tejidos y fluidos corporales148. Cruza la barrera placentaria. Su concentración en la leche materna igual a la plasmática.
La vida media con función renal normal es de 1 a 4 horas, siendo en los acetiladores rápidos de 0,5-1,6 horas y en los lentos de 2-5 horas149. En pacientes con alteración de la función hepática o renal la VM se prolonga a 4,3 horas.
Su metabolismo es principalmente hepático a través de una acetilación (genéticamente determinada) que produce acetilisoniacida. Posteriormente son hidrolizados a ácido isonicotínico y a acetilhidracina. Se cree que uno de los metabolitos de esta acetilhidracina es el responsable de la hepatotoxicidad inducida por INH.
Hasta un 70% se excreta por orina, en caso de función renal inalterada y tras una dosis de 5 mg/Kg, aunque pueden existir diferencias entre los metabolitos eliminados en función del tipo de acetilador150.
Mecanismo de toxicidad
La INH ejerce su acción toxicológica al producir alteraciones en el metabolismo de la piridoxina, reduciendo los depósitos de la misma. Se produce un incremento la excreción renal de piridoxina a través de la formación de hidrazonas piridoxina-INH. Las hidrazonas actúan competitivamente con la enzima piridoxina kinasa, la cual es la encargada de transformar la piridoxina en su forma fisiológicamente activa que es el fosfato de piridoxina. La piridoxina a su vez es un cofactor en la producción del neurotransmisor inhibidor GABA (ácido gamma-aminobutírico). El resultado final en una sobredosis de INH es la depleción del GABA y como consecuencia una desinhibición supraespinal, lo cual explicaría las convulsiones en la intoxicación por INH.
La acidosis metabólica que presentan estos pacientes se debe a la propia acidosis láctica producida por las convulsiones, a un bloqueo en la conversión de lactato a piruvato, y a una alteración en el metabolismo de la glucosa con un aumento en el metabolismo de los ácidos grasos, produciéndose hiperglucemia y cetonuria151.
Diferentes fármacos pueden interactuar con la INH aumentando o disminuyendo sus niveles al actuar sobre el proceso absortivo o sobre los mecanismos metabólicos. La asociación concomitante de la INH con otras drogas puede producir respuestas psiquiátricas (antidepresivos tricíclicos), hiperglucemia (clorpropamida), agitación o temblor parkinsoniano (levodopa), hipotensión (meperidina), incremento de la hepatotoxicidad (rifampicina), hipertensión, taquicardia e hipertermia (inhibidores de la monoaminooxidasa).
Clínica
Las manifestaciones tóxicas se suelen manifestar de 1-2 horas tras la ingesta, con un rango de presentación de 30 minutos hasta 7 horas. Los efectos tóxicos en una primera fase incluyen naúseas, vómitos, visión borrosa, visión de luces de colores, vértigos y dificultad para hablar. Rápidamente acontece una segunda fase con convulsiones tónico-clónicas refractarias a tratamiento, respiración dificultosa, coma y una profunda acidosis metabólica.
Otros síntomas que pueden estar presentes incluyen: hipertermia, taquicardia, hipotensión, taquipnea con respiración de Kussmaul, rabdomiolisis152. No está descrita una cardiotoxicidad directa, pero puede producirse parada cardíaca debido a severa hipoxemia y/o acidosis metabólica.
Diagnóstico diferencial
Incluye las sustancias que pueden producir convulsiones tóxicas y anión gap elevado: organofosforados, teofilina, isoniacida, insulina; simpaticomiméticos, salicilatos. Cocaína, antidepresivos, metilxantinas, phencyclidina, betabloqueantes, etanol (síndrome de abstinencia), litio, lidocaína , lindano.
Tratamiento
Si el paciente está asintomático 6 horas postingestión, es poco probable que desarrolle complicaciones. No se debe intentar provocar el vómito, ya que las convulsiones pueden aparecer de forma repentina, con riesgo de aspiración bronquial.
El control de las convulsiones se hará con diazepan. Debemos corregir la acidosis metabólica si pH<7,2. El fallo respiratorio se tratará con oxigenoterapia y/o intubación, y la corrección de la hipotensión con expansores plasmáticos y/o drogas vasoactivas153.
Si la ingesta ha sido hace menos de una hora, realizar lavado. Si la ingesta se ha producido hace unas pocas horas, administrar una dosis de carbón activado (1-2 g/Kg)154.
El antídoto específico es la piridoxina155. En los paciente con convulsiones de origen desconocido debería usarse de forma empírica la piridoxina junto con benzodiacepinas (tienen efecto sinérgico). La fenitoína en estos casos no es efectiva. La dosis del antídoto es de un gramo de piridoxina por cada gramo de INH ingerida (durante cinco minutos). Es caso de dosis tóxica desconocida administrar 5 gramos de piridoxina durante cinco minutos. En caso de que las convulsiones no se controlen después de administrar 10 gramos de piridoxina, es preciso reconsiderar el diagnóstico156.
En caso de rabdomiolisis mantener una adecuada diuresis y alcalinización urinaria.
Evolución y pronóstico
El pronóstico depende de la cantidad ingerida y de la precocidad del tratamiento. Con adecuado tratamiento y en ausencia de complicaciones el pronóstico es normalmente bueno después de 24-48 horas.
La muerte en estadios precoces (1-12 horas) es debida al fallo respiratorio y/o parada cardiaca (acidosis, hipoxemia) como resultado del status convulsivo.
BIBLIOGRAFÍA
1. Burillo-Putze G, Munne P, Dueñas A, Pinillos MA, Naviero JM, Cobo J et al. National multicenter study of acute intoxication in Emergengy Departments of Spain. Eur J Emerg Med 2003 (en prensa). [ Links ]
2. Mintegui Raso S, Benito Fernández J, Vázquez Ronco MA, Fernández Landulce A, Gortázar Arias P, Grau Bolado G. Intoxicaciones en urgencias: cambios epidemiológicos en los últimos 10 años. Rev Esp Pediatr 2002; 56: 23-29. [ Links ]
3. Divoll M, Greenblatt DJ, Ameer B, Abernety DR. Effect of food on acetaminophen absorption in young and elderly subjects. J Clin Pharmacol 1982; 22: 571-576. [ Links ]
4. Caraccio TR, Mofenson HC, Lawlles MJ. Delayed peak acetaminophen (APAP) levels (abstract). J Toxicol Clin Toxicol 1997; 35: 563. [ Links ]
5. Baselt RC. Disposition of Toxic Drugs and Chemicals in Man. 5th ed. California: Chemical Toxicology Institute, 2000. [ Links ]
6. Corcoran GB, Mitchel JR, Vaishnav YN, Horning EC. Evidence that acetaminophen and N-hydroxiacetaminophen from a common arylating intermediate. N-acetyl-p-benzoquinoneimine. Mol Pharmacol 1980; 18: 536-542. [ Links ]
7. Clissold SP. Paracetamol and phenacetin. Drugs 1986: 32 S4: 46-59. [ Links ]
8. Prescott LF. Paracetamol overdosage: pharmacological considerations and clinical management. Drugs 1983; 25: 290-314. [ Links ]
9. Corcoran GB, Mitchel JR. Evidence for redox cycling of acetaminophen and its reactive metabolite by endogenous microsomal systems. Adv Exp Med Biol 1981;136 Pt B:1085-1098. [ Links ]
10. Mitchel JR, Jollow DJ, Potter WZ. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther 1973; 187: 185-194. [ Links ]
11. Hoivik DJ, Manautou JE, Tviet A, Mankowski DC, Khairallah EA, Cohen SD. Gender-related differences in susceptibility to acetaminophen-induced protein arylation and nephrotoxicity on the CD-1 mouse. Toxicol Appl Pharmacol 1995; 130: 257-271 [ Links ]
12. Sakaida I, Kayano K, Wasaki S, Nagatomi A, Matsumura Y, Okita K. Protection against acetaminophen-induced liver injury in vivo by an iron chelator, deferoxamine. Scand J Gastroenterol 1995; 30: 61-67. [ Links ]
13. Jaeschke H, Mitchell JR. Neutrophil accumulation exacerbates acetaminophen-induced liver injury. (Abstract 4031) FASEB J 1989; A920. [ Links ]
14. Tirmenstein MA, Nelson SD. Subcellular brinding and effects on calcium homeostasis produced by acetaminophen and a non-hepatotoxic regioisomer, 3 hydroxyacetoanilide in mouse liver. J Biol Chem 1989; 264: 9814-9819. [ Links ]
15. Donnelly PG, Walker RN, Racz WJ. Inhibition of mitochondrial respiration in vivo is an early event in acetaminophen-induced hepatotoxicity. Arch Toxicol 1994; 68: 110-118. [ Links ]
16. Whyte IM, Buckley NA, Reith D, Goodhew I, Seldon M, Dawson AH. Acetaminophen causes an increased international normalized ratio by reducing functional factor VII. Ther Drug Monitor 2000; 22: 742-748. [ Links ]
17. Lawrence IG, Lear J, Burden AC, Maddocks E, Smith JF. Hyperglicemia induced by paracetamol. Postgrad Med J 1995; 71:702. [ Links ]
18. Yang CC, Deng JF, Lin TJ. Pancytopenia, hyperglycemia, shock, coma, rhabdomyolisis, and pancreatitis associated with acetaminophen poisoning. Vet Human Toxicol 2001; 43: 344-348. [ Links ]
19. Eguia L, Materson BJ. Acetaminophen-related acute renal failure without fulminant liver failure. Pharmacotherapy 1997; 17: 363-370. [ Links ]
20. Prescott LF, Proudfoot AT, Cregeen RJ. Paracetamol-induced acute renal failure in the absence of fulminant liver damage. Br J Med (Clin Res Ed) 1982; 284: 421-422. [ Links ]
21. Rumack BH. Acetaminophen overdose in children and adolescents. Pediatr Clin North Am 1986; 33: 691-701. [ Links ]
22. Cheung L, Potts RG, Meyer KC. Acetaminophen treatment nomogram. N Engl J Med 1994; 330: 1907-1908. [ Links ]
23. Thummel KE, Slattery JT Ro H, Chien JY, Nelson SD, Lown KE, Watkins PB. Ethanol and production of the hepatotoxic metabolite of acetaminophen in healthy adults. Clin Pharmacol Ther 2000; 67: 591-599. [ Links ]
24. Rumack BH. Acetaminophen overdose in young children: Treatment and effects of alcohol and other additional ingestants in 417 cases. Am J Dis Child 1984; 138:428-433. [ Links ]
25. Murphy R, Swartz R, Watkins PB. Severe acetaminophen toxicity in a patient receiving isoniazid. Ann Intern Med 1990; 113: 799-800. [ Links ]
26. Buckley NA, Whyte IM, OConnell DL. Activated charcoal reduces the need for N-acetylcysteine treatment after acetaminophen (paracetamol) overdose. J Toxicol Clin Toxicol 1999; 37: 753-757. [ Links ]
27. Neuvonen PJ, Vartiainen M, Tokola O. Comparison of activated charcoal and ipecac syrup in prevention of drug absorption. Eur J Clin Pharmacol 1983; 24: 557-562. [ Links ]
28. Spiller HA, Krenzelok EP, Grande GA. A prospective evaluation of the effect of activated charcoal before oral N-acetylcisteine on indirect indicators of tissue oxygenation in septic shock patients. Crit Care Med 1994; 22: 1738-1746. [ Links ]
29. Caravati EM. Unintentional acetaminophen ingestion in children and the potential for hepatotoxicity. J Toxicol Clin Toxicol 2000; 38: 291-296. [ Links ]
30. Lauterburg BH, Corcoran GB, Mitchel JR. Mechanism of action of N-acetylcysteine in the protection against hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71: 980-991. [ Links ]
31. Buckpitt AR, Rollins DE, Mitchel JR. Varying effects sulfhydryl nucleophiles on acetaminophen oxidation and sulfhydryl adduct formation. Biochem Pharmacol 1979; 28: 2941-2946. [ Links ]
32. Slattery JT, Wilson JM, Kalhorn TF, Nelson SD. Dose-dependent pharmacokinetics of acetaminophen: Evidence for glutathione depletion in humans. Clin Pharmacol Ther 1987; 41: 413-418. [ Links ]
33. Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55: 871-876. [ Links ]
34. Bridger S, Henserson K, Glucksman E, Ellis AJ, Henry JA, Williams R. Lesson of the week, Deaths from low-dose paracetamol poisoning. Br Med J 1998; 316:1724-1725. [ Links ]
35. Smilkstein MJ, Bronstein AC, Linden C, Augenstein WL, Kulig KW, Rumack BH. Acetaminophen overdose: a 48-hour intravenous n-acetylcysteine treatment protocol. Ann Emerg Med 1991; 20: 1058-1063. [ Links ]
36. Keays R, Harrison PM, Wendon JA, Forbes A, Gove C, Alexander GJ, Williams R. Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: A prospective controlled trial. Br Med J 1991; 303:1026-1029. [ Links ]
37. Tenenbein PK, Sitar DS, Tenenbein M. Interaction between N-acetylcysteine and activated charcoal: implications for the treatment of acetaminophen poisoning. Pharmacother 2001; 21: 1331-1336. [ Links ]
38. Krenzeiok EP. Use of activated charcoal. Ann Emerg Med 1986; 15: 102-103. [ Links ]
39. Smilkstein MJ, Rumach BH. Elimination halflife as a predictor of acetaminophen-induced hepatotoxicity (abstract). Vet Human Toxicol 1994; 36: 377. [ Links ]
40. Woo OF, Mueller PD, Olson KR, Anderson IB, Kim SY. Shorter duration of n-acetylcysteine (NAC) for acute acetaminophen overdose. Ann Emerg Med 2000; 35: 363-368. [ Links ]
41. Clark D, Ruck B, Jennis T. Compliance with PCC recommendations : discontinuation of NAC (abstract) J Toxicol Clin Toxicol 2001; 39: 485. [ Links ]
42. Bernal W, Donaldson N, Wyncoll D, Wendon J. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: a cohort study. Lancet 2002; 359: 558-563. [ Links ]
43. Gilman AG, Goodman LS, Gilman A. Pharmacological basis of therapeutics, 6th ed. New York, MacMillan Co. 1980. [ Links ]
44. Hill JB. Experimental salicylate poisoning: observations on the effects of altering blood pH on tissue and plasma salicylate concentrations. Pediatrics 1971;47: 658-665. [ Links ]
45. Levy G, Leonards JR. Urine pH and salicylate therapy. JAMA 1971; 217: 81. [ Links ]
46. Thurston GH, Pollock PG, Warren SK. Reduced brain glucose with normal plasma glucose in salicylate poisoning. J Clin Invest 1970; 49: 2139-2145. [ Links ]
47. Montgomery H, Porter JC, Bradley RD. Salicylate intoxication causing a severe systemic inflammatory response and rhaddomyolisis Am J Emerg Med 1994; 12: 531-532. [ Links ]
48. Cazals Y. Auditory sensori-neural alterations induced by salicylate. Progr Neurobiol 2000 ; 62: 583-681. [ Links ]
49. Krause DS, Wolf BA, Shaw LM. Acute aspirin overdose: mechanisms of toxicity. Ther Drug Monit 1992; 14: 441- 451. [ Links ]
50. Watson JE, Tagupa ET. Suicide attempt by means of aspirin enema. Ann Pharmacother 1994; 28: 467- 469. [ Links ]
51. Goldfrank LR, WeismanRS, Flomenbaum NE. Goldfrank s Toxicologic Emergencies, 5th ed. Appleton & Lange, Norwalk CT, 1994. [ Links ]
52. AACT y EAPCCT. Position statement and practice guidelines on the use of multi-dose activated charcoal in the treatment of acute poisoning. J Toxicol Clin Toxicol 1999; 37: 731-751. [ Links ]
53. Fillippone G, Fish S, Lacouture P. Reversible adsorption (desadsorption) of aspirin from activated charcoal. Arch Intern Med 1987; 147: 1390-1392. [ Links ]
54. Prescott LF, Balali-Mood M, Critchley JA, Johnstone AF, Proudfoot AT. Diuresis or urinary alkalinization for salicylate poisoning? Br Med J 1982; 285:1383-1386. [ Links ]
55. Proudfoot A. Toxicity of salicylates. Am J Med 1983; 75: 99-103. [ Links ]
56. Jacobsen D, Wiik-Larsen E, Bredesen JE. Haemodalysis or haemoperfusion in severe salicylate poisoning. Human Toxicol 1988; 7: 161-163. [ Links ]
57. Pinkston R, Walker LA. Multiorgan system failure caused by valproic acid toxicity. Am J Emerg Med 1997; 15: 504-506. [ Links ]
58. Post RM. Issues in the long-term management of bipolar affective illness. Psychiatr Ann 1993; 23: 86-93. [ Links ]
59. Mathew NT, Kailasam J, Meadors L, Chernyschev O, Gentry P, Intravenous Valproate Sodium (Depacon) Aborts Migraine Rapidly: A Preliminary Report. Headache 2000; 40: 720-723. [ Links ]
60. Norton J. Use of intravenous valproate sodium in status migraine. Headache 2000; 40: 755-757. [ Links ]
61. Davis LL, Ryan W, Adinoff B, Petty F. Comprehensive review of the psychiatric uses of valproate. J Clin Psychopharmacol 2000; 20 (Suppl. 1): 1S–7S. [ Links ]
62. Litovitz TL, Bailey KM, Schmitz BF, Holm KC, Klein-Schwartz W. 1990 Annual report of the American Association of Poison Control Centers National Data Collection System. Am J Emerg Med 1991; 9: 461–509. [ Links ]
63. Litovitz TL, Klein-Schwartz W, White S, Cobaugh DJ, Youniss J et al. 2000 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med 2001; 19: 337-395. [ Links ]
64. Klotz U, Antonin KH. Pharmacokinetics and bioavailability of sodium valproate. Clin Pharmacol Ther 1977; 21: 736-743. [ Links ]
65. Gugler R, von Unruh GE. Clinical pharmacokinetics of valproic acid. Clin Pharmacol 1980; 5: 67-83. [ Links ]
66. Duarte J, Macias S, Coria F, Fernandez E, Claveria LE. Valproate-induced coma: Case report and literature review. Ann Pharmacother 1993; 27: 582-583. [ Links ]
67. Kane SL, Constantiner M, Staubus AE, Meinecke CD, Sedor JR. High-Flux Hemodialysis without hemoperfusion is effective in acute valproic acid overdose. Ann Pharmacother 2000; 34: 1146-1151. [ Links ]
68. Chadwick DW. Concentration-effect relationships of valproic acid. Clin Pharmacol 1985; 10: 155-163. [ Links ]
69. Kandrotas RJ, Love JM, Gal P, Oles KS. The effect of hemodialysis and hemoperfusion on serum valproic acid concentration. Neurology 1990; 40: 1456-1458. [ Links ]
70. Kingsley E, Tweedale R, Gray P. The role of toxic metabolites in the hepatotoxicity of valproic acid. Gastroenterology 1980; 79: 511-518. [ Links ]
71. Pinder PM, Borgden RN, Speight TM. Sodium valproate: A review of its pharmacological properties and therapeutic efficacy in epilepsy. Drugs 1977; 13: 81-92. [ Links ]
72. Nau H, Loscher W. Valproic acid and metabolites: pharmacological and toxicological studies. Epilepsia 1984; 25 (S1): S14-S22. [ Links ]
73. Albus H, Williamson R. Electrophysical analysis of the action of valproate on pyramidal neurons in the rat hippocampal slice. Epilepsia 1998; 39: 124-139. [ Links ]
74. Mortensen PB, Hansen HE, Pederson B, Hartmann-Andersen F, Husted SE. Acute valproate intoxication: Biochemical investigations and hemodialysis treatment. Int J Clin Pharmacol Ther Toxicol 1983; 21: 64- 68. [ Links ]
75. Graudins A, Aaron C.K. Delayed peak serum valproic acid in massive divalproex overdose-Treatment with charcoal hemoperfusion. J Toxicol Clin Toxicol 1996; 34: 335-341. [ Links ]
76. Ingels M, Johnson C, Rangan C, Cantrell L, Beauchamp J, Clark RF. Retrospective study of delayed valproic acid toxicity. Acad Emerg Med 2000; 7: 499. [ Links ]
77. Johnson LZ, Martinez I, Fernandez MC, Davis CP, Kasinath BS Successful treatment of valproic acid overdose with hemodialysis. Am J Kidney Dis 1999; 33: 786-789. [ Links ]
78. Spiller HA, Krenzelok EP, Klein-Schwartz W, Winter ML, Weber JA, Sollee DR et al. Multicenter case series of valproic acid ingestion: serum concentrations and toxicity. J Toxicol Clin Toxicol 2000; 38: 755- 760. [ Links ]
79. Andersen G, Ritland S. Life threatening intoxication with sodium valproate. J Toxicol Clin Toxicol 1995; 33: 279-284. [ Links ]
80. Khoo SH, Leyland MJ. Cerebral edema following acute sodium valproate overdose. J Toxicol Clin Toxicol 1992; 30: 209-214. [ Links ]
81. Dupuis RE, Lichtman SN, Pollack GM. Acute valproic acid overdose. Clinical course and pharmacokinetic disposition of valproic acid and metabolites. Drug Saf 1990; 5: 65-71. [ Links ]
82. Loscher W, Nau H. Pharmacological evaluation of various metabolites and analogues of valproic acid. Anticonvulsant and toxic potencies in mice. Neuropharmacology 1985; 24: 427- 435. [ Links ]
83. Hertz L, Yu ACH, Kala G, Schousboe A, Neuronal-astrocytic and cytosolic-mitochondrial metabolite trafficking during brain activation, hyperammonemia, and energy deprivation. Neurochem Int 2000; 37: 83-102. [ Links ]
84. Camfield PR, Bagnell P, Camfield CS, Tibbles JA. Pancreatitis due to valproic acid. Lancet 1979; 1: 1198–1199. [ Links ]
85. Cotariu D, Zaidman JL. Valproic acid and the liver. Clin Chem 1988; 34: 890-897. [ Links ]
86. Caparros-Lefebvre D, Lecomte-Houcke M, Pruvot FR, Declerck N, Paris JC, Petit H. Unusual electronmicroscopic changes in valproate-associated liver failure. Lancet 1993; 341: 1604. [ Links ]
87. Beauchamp J, Olson K. Valproic acid overdoses: A retrospective study comparing serum drug levels and the incidence of adverse outcomes. J Toxicol Clin Toxicol 1999; 37: 637-638. [ Links ]
88. Tank JE, Palmer BF. Simultaneous in series hemodialysis and hemoperfusion in the management of valproic acid overdose. Am J Kidney Dis 1993; 22: 341-344. [ Links ]
89. Ruskoky D, Holmes C, Schauben J. Utilization of hemodialysis to enhance valproic acid (VPA) elimination. J Toxicol Clin Toxicol 1999; 37: 636. [ Links ]
90. Steiman GS, Woerpel RW, Sherard ES. Treatment of accidental sodium valproate overdose with an opiate antagonist. Ann Neurol 1979; 6: 274. [ Links ]
91. Mortensen PB, Hansen HE, Pedersen B, Hartmann-Andersen F, Husted SE. Acute valproate intoxication: biochemical investigations and hemodialysis treatment. Int J Clin Pharmacol Ther Toxicol 1983; 21: 64-68. [ Links ]
92. Sztajnkrycer MD, Seaglione JM, Bond GR. Valproate-induced hyperammonemia: Preliminary evaluation of ammonia elimination with carnitine administration. J Toxicol Clin Toxicol 2001; 39: 497. [ Links ]
93. Litovitz TL, Normann SA, Veltri JC. 1985 annual report of the American Association of Poison Control Centers National Data Collection System. Am J Emerg Med 1986; 4: 427-458. [ Links ]
94. Sumner DJ, Kalk J, Whiting B. Metabolism of barbiturate after over-dosage. Br Med J 1975; 1: 335. [ Links ]
95. Ellenhorn MJ. Barbiturates. En Ellenhorn MJ, Barceloux DG (eds): Medical Toxicology: Diagnosis and Treatment of Human Poisoning. New York, Elsevier, 1988: 576. [ Links ]
96. McCarron MM, Schulze BW, Walberg CB, Thompson GA, Ansari A. Short-acting barbiturate overdosage. Correlation of intoxication score with serum barbiturate concentration. JAMA 1982; 248: 55-61. [ Links ]
97. Carroll BJ. Barbiturate overdosage: Presentation with focal neurological signs. Med J Aust 1969; 1: 1133- 4135. [ Links ]
98. Yatzidis H. Bullous lesions in acute barbiturate poisonings. JAMA 1971; 217: 211. [ Links ]
99. Wilber GS, Coldwell BB, Trenholm HL. Toxicity of ethanol-barbiturate mixtures. J Pharm Pharmacol 1969; 21: 232. [ Links ]
100. Pond SM, Lewis-Driver DJ, Williams, GM, Green AC, Stevenson NW. Gastric emptying in acute overdose : A prospective randomized controlled trial. Med J Aust 1995; 163: 345-349. [ Links ]
101. Holzer P, Beubler B, Dirnhofer R. Barbiturate poisoning and gastrointestinal propulsion. Arch Toxicol 1987; 60: 394-396. [ Links ]
102. Wakabayashi Y, Maruyama S, Hachimura K, Ohwada T. Activated charcoal interrupts enteroenteric circulation of Phenobarbital. J Toxicol Clin Toxicol 1994; 32: 419-424. [ Links ]
103. Zawada ET, Nappi J, Done G, Roliins D. Advances in the hemodialysis management of phenobarbital overdose. South Med J 1983; 76: 6-8. [ Links ]
104. Graves NM, Brundaje RC, Wen Y. Population pharmacokinetics of carbamazepine in adults with epilepsy. Pharmacotherapy 1998;18:273-281. [ Links ]
105. Sweetman S (ed). Carbamazepine. En: Martindale. The Complete Drug Reference. London: Pharmaceutical Press, 2001. [ Links ]
106. Weaver DF, Camfield P, Frazer A. Massive carbamazepine overdose: clinical and pharmacologic observation in five episodes. Neurology 1988; 38: 755-759. [ Links ]
107. Westenberg HGM, Klein E van der, Oei TT, Zeeuw RA. Kinetics of carbamazepine and carbamazepine epoxide determined by use of plasma and saliva. Clin Pharmacol Ther 1978; 23: 320-328. [ Links ]
108. Patsalos PN, Krishna S, Elyas AA. Carbamazepine and carbamazepine 10,11 epoxide pharmacokinetics in an overdose patient. Hum Toxicol 1987; 6: 241-244. [ Links ]
109. Yacobi A, Zlotnick S, Colaizzi JL. A multiple-dose safety and bioequivalence study of a narrow therapeutic index drug: A case for carbamazepine. Clin Pharmacol Ther 1999; 65: 389-394. [ Links ]
110. Lingamaneni R, Hemmings HC. Effects of anticonvulsants on veratridine and KCL evoked glutamate release from rat cortical synaptosomes. Neurosci Lett 1999; 276: 127-130. [ Links ]
111. Yoshimura R, Yanagihara N, Terao T, Minami K, Abe K, Izumi F. Inhibition by carbamazepine of various ion channels mediated catecholamine secretion in cultured bovine adrenal medullary cells, Naunyn-Schmiedebergs. Arch Pharmacol 1995; 352: 297-303. [ Links ]
112. Skerritt JH, Davies LP, Johnston GA. Interactions of the anticonvulsant carbamazepine with adenosine receptors. Epilesia 1983; 24: 634-642. [ Links ]
113. Lampe H, Bigalke H. Carbamazepine blocks NMDA-activated currents in cultured spinal cord neurons. Neuroreport 1990; 1: 26-28. [ Links ]
114. Salcman M, Pippenger CE. Acute carbamazepine encephalopaty. J Am Med Assoc 1975; 231: 915. [ Links ]
115. Lehrman SN, Bauman ML. Carbamazepine overdose. Am J Dis Child 1981; 135: 768-769. [ Links ]
116. Beerman B, Edhag O, Vallin H. Advanced hearth block aggravated by carbamazepine. Br Heart J 1975; 37: 668. [ Links ]
117. Stevens WP. Water intoxication due to carbamazepine. Br Med J 1977; 1:754. [ Links ]
118. Nilsson C, Sterner G, Idvall J. Charcoal hemoperfusion for treatment of serious carbamazepine poisoning. Acta Med Scand 1984; 216: 137-140. [ Links ]
119. Drenck NE, Risbo A. Carbamazepine pisoning, a surprisingly severe case. Anaesth Intensive Care 1980; 8: 203-205. [ Links ]
120. Sullivan JB, Rumack BH, Peterson RG. Acute carbamazepine toxicity resulting from overdose. Neurology 1981;31:621-624. [ Links ]
121. de Zeeuw RA, Westenberg HG, van der Kleijn E, Gimbrere JS. An unusual case of carbamazepine poisoning with a near-fatal relapse after two days. Clin Toxicol 1979;14: 263-269. [ Links ]
122. Sethna M, Solomon G, Cedarbaum J, Kutt H. Succesful treatment of massive carbamazepine overdose. Epilepsia 1989; 30: 71-73. [ Links ]
123. Bates N, Edwards N, Roper J, Volans G (eds). Paediatric Toxicology. Hadbook of Poisoning in Children. London, Macmillan Reference Ltd, 1997. [ Links ]
124. Hojer J, Malmlund H, Berg A. Clinical features in 28 consecutive cases of laboratory confirmed massive poisoning with carbamazepine alone. Clin Toxicol 1993; 31: 449-458. [ Links ]
125. Durelli L, Massazza V, Cavallo R. Carbamazepine toxicity and poisoning. Incidence, clinical features and management. Med Toxicol Adv Drug Experience 1989; 4: 95-107. [ Links ]
126. Schmidt S, Schmitz-Buhl M. Signs and symptoms of carbamazepine overdose. J Neurol 1995; 242: 169- 173. [ Links ]
127. Lacroix J, Gaudreault P, Gauthier M. Admission to a pediatric intensive care unit for poisoning: A review of 105 cases. Crit Care Med 1989; 17: 748-750. [ Links ]
128. Kasarskis EJ, Kuo C-S, Berjer R, Nelson KR. Carbamazepine-induced cardiac dysfunction: characterization of two distinc clinical syndromes. Arch Intern Med 1992; 152: 186- 191. [ Links ]
129. Tsao CY, Wright FS. Acute chemical pancreatitis associated with carbamazepine intoxication. Epilepsia 1993; 34: 174-176. [ Links ]
130. Edge W, Edmonds J. Serum sodium and carbamazepine overdose. Clin Toxicol 1992; 30: 479- 480. [ Links ]
131. MacNab AJ, Birch P, MacReady J. Carbamazepine poisoning in children. Pediatr Emerg Care 1993; 9: 195-198. [ Links ]
132. Seymour JF. Carbamazepine overdose: Features of 33 cases. Drug Safety 1993; 8: 81-88. [ Links ]
133. Wason S, Baker RC, Carolan P, Seigel R, Druckenbrod RW. Carbamazepine overdose-the effects of multiple dose activated charcoal. Clin Toxicol 1992; 30: 39-48. [ Links ]
134. Deshpande G, Meert KL, Valentini RP. Repeat charcoal hemoperfusion treatments in life threatening carbamazepine overdose. Pediatr Nephrol 1999; 13: 775-777. [ Links ]
135. McNamara JO. Drugs effectivive in the therapy of the epilepsies. En: Hardman JG, Limbird LE, Molinoft PB et al (eds): Goodman and Gilman´s The Pharmacological Basis of Therapeutics. 9 th ed, New York, McGraw-Hills, 1996; 468. [ Links ]
136. Helfant TE, Seuffer GW, Patton RD, Stein E, Damato AN. The clinical use of difenil-hydantoin (Dilantin) in the treatment and prevention of cardiac arrhytmias. Am Heart J 1969; 77: 315- 323. [ Links ]
137. Dona S. Phenytoin and other anticonvulsant. Hadda Lester M, Winchester James F. Philadelphia. W.B. Saunders: 1990; 877-893. [ Links ]
138. Luef G, Burtscher J, Kremser C, Birbamer G, Aichner F, Bauer G et al. Magnetic resonance volumetry of the cerebellum in epileptic patients after phenytoin overdosages. Eur Neurol 1996; 36: 273-277. [ Links ]
139. Chaikin P, Adir J. Unusual absorption profile of phenytoin in a masive overdose case. J Clin Pharmacol 1987; 27: 70-73. [ Links ]
140. Patel H, Crichton JU. The neurologic hazards of diphenilhydantoina in childhood. J Pediatr 1968; 73: 676-684. [ Links ]
141. Borosfsky LG, Louis S, Kutt H, Roginsky M. Diphenylhydantoin efficacy, toxicity and dose-serum relationships in children. J Pediatr 1972; 81: 995-1002. [ Links ]
142. HC Chua, Venketasubramanian N, Tan CB, Tjia H. Paradoxical seizures in phenytoin toxicity. Singapore Med J 1999; 40: 230-233. [ Links ]
143. Curtis DL, Piibe R, Ellenhorn MJ, Wasserberg J, Ordog G. Phenytoin toxicity: predictors of clinical course. Vet Hum Toxicol 1989: 31: 162-163. [ Links ]
144. Stilman N, Masdeu SC. Incidence of seizures with phenytoin toxicity. Neurology 1985; 35: 1769-1772. [ Links ]
145. Zoneraich S, Zoneraich O, Siegel J. Sudden death following intravenous sodium diphenylhydantoin. Am Heart 1976; 91: 375-377. [ Links ]
146. Jacobsen D, Alvik A, Bresesen JE. Pharmacokinetics of phenytoin. Acute intoxication in an adult and two children. (Abst). Vet Hum Toxicol 1986; 55: 412-413. [ Links ]
147. Boxenbaum HG, Riegelman S. Determination of isoniazid and metabolites and biological fluids. J Pharm Sci 1974; 63: 1191- 1197. [ Links ]
148. Kucers A, Bennet N. The use of antibiotics. 3rd ed. London; Williams Heinemann Medical Book Ltd, 1979. [ Links ]
149. Ellard GA. The potential clinical significance of the isoniazid acetylator phenotype in the tratment of pulmonary tuberculosis. Tubercle 1984; 65: 211-227. [ Links ]
150. Wood JD, Peesker. A correlation between changes in GABA metabolism and isonicotinic acid hydrazine-induced seizures. Brain Res 1972; 45: 489-498. [ Links ]
151. Terman DS, Teitelbaum DT. Isoniazid sef poisoning. Neurology 1970; 20: 299-304. [ Links ]
152. Davies DM. Textbook of adverse drug reactions, 2nd ed. New York, Oxford University Press, 1981. [ Links ]
153. Orlowsky JP, Paganini EP, Pippenger CE. Treatment of a potentially lethal dose isonizide ingestion. Annals Emerg Med 1988; 17: 73-76. [ Links ]
154. Siefkin AD, Alberston TE, Corbett MG. Isoniazid overdose: pharmackinetics of oral charcoal in treatment. Hum Toxicol 1987; 6: 497-501. [ Links ]
155. Watson S, Lacouture PG, Lovejoy FH. Single highdose pyridoxine treatment for isoniazid overdose. JAMA 1981; 246: 1102-1104. [ Links ]
156. Stewart MF, Freemont AJ, Richardson T. Fatal isoniazid poisoning. Ann Clin Biochem 1995; 32: 229-231. [ Links ]
Anexo 1. Criterios para indicar de forma correcta la administración de NAC en distintas situaciones clínicas.
A. INTERVALO 0-16 h post-ingesta (corresponde a la inmensa mayoría de casos) 1. La dosis afirmada o calculada es tóxica Deben de cumplirse dos condiciones: disponer de una anamnesis altamente fiable y sobredosis de paracetamol dentro del intervalo de toxicidad (> 125-150 mg/Kg en adultos-adolescente ó > 200 mg/Kg en niños menores de 6 años) Si se cumplen las dos condiciones se procederá a: Solicitar paracetamolemia esperando a las 4 h post-ingesta en los casos atendidos precozmente o lo antes posible en los casos atendidos con posterioridad.
Empezar la administración de NAC en los casos con un intervalo asistencial mayor de 8 h o cuando se prevé que el resultado analítico no estará disponible hasta transcurridas 8 h post-ingesta.
Si el resultado analítico está por debajo de la línea de riesgo según el nomograma 150 no se administrará NAC o se suspenderá si se había iniciado y se procederá al alta toxicológica.
Si el resultado analítico está por encima de la línea de riesgo según el nomograma 150 se continuará la NAC (si se hubiera iniciado) o se empezará su administración.
Si no se dispone de paracetamolemia urgente, se administrará el antídoto NAC.
Valorar los pacientes con un mayor riesgo hepatotóxico para en su caso reducir los valores del nomograma 150.2. La dosis afirmada o calculada es subtóxica Este supuesto sólo debe aceptarse bajo dos condiciones: disponer de una anamnesis altamente fiable o la sobredosis de paracetamol calculada está por debajo de los límites de toxicidad.
Si se cumplen las dos condiciones, se procederá a: Alta toxicología. Control ambulatorio a las 12-24 h.
Ante cualquier duda, actuar como en el supuesto A 1.3. La dosis de paracetamol no puede averiguarse o no puede hacerse con fiabilidad Se procederá como en el supuesto A 1 (dosis tóxica). B. INTERVALO 16-24 h post-ingesta Debe procederse de igual modo que en el apartado A (intervalo 0-16 h.) o sea indicando NAC o alta toxicológica según la dosis ingerida y la paracetamolemia.
A pesar de estos criterios de actuación similar, este intervalo (16-24 h) presenta unas peculiaridades específicas: De entrada, debe empezarse la administración de NAC a menos que la dosis calculada o afirmada de paracetamol sea claramente atóxica. En tal caso se procede al alta.
Si la sobredosis está en el rango tóxico, debe solicitarse una paracetamolemia.
Si es positiva será indicación (en función del nomograma) de suspender o continuar la NAC ya iniciada.
Si la paracetamolemia es negativa no debe comportar la suspensión de la NAC ya que puede tratarse de un falso negativo debido a que en este intervalo, en general, las concentraciones de paracetamol son del orden de 10 mg/ml o inferiores, cifra que está por debajo de los límites de detección del método analítico usado habitualmente en nuestro medio.
Por lo tanto una paracetamolemia negativa debe continuarse la NAC hasta transcurridas 24-36 h post-ingesta suspendiéndola si se comprueba que no hay citolisis hepática. En caso contrario, debe continuarse.
Debe solicitarse siempre unas GOT/GPT ya que su aumento por sí mismo indica que es preciso administrar la NAC con independencia de otros factores.C. INTERVALO > 24 h En general, la paracetamolemia no es detectable en los casos asistidos dentro de este intervalo pero aunque lo fuera (debido a una importante sobreingesta que alarga la vida media del paracetamol) no podría valorarse en el nomograma 150 ya que este carece de fiabilidad transcurridas 24 o más horas desde la ingesta.
Así pues, en los casos de latencia larga, la indicación de administrar NAC no se basará en el nomograma sino en otros parámetros.
Se procederá de la siguiente forma:1. La dosis calculada o afirmada es subtóxica
Si la dosis subtóxica se basa en una anamnesis totalmente fiable, se puede dar el alta toxicológica. Es conveniente comprobar que las GOT/GPT son normales para asegurar que el alta médica es correcta. En caso de elevación de las GOT/GPT actuar como en C2.2. La dosis calculada o afirmada es tóxica o no puede definirse
En caso de dosis no definida o dosis en el rango tóxico, se actuará de la siguiente forma: Si las GOT/GPT están elevadas con independencia de la paracetamolemia, administrar NAC.
Si las GOT/GPT son normales y la concentración sérica de paracetamol es detectable, es prudente iniciar la NAC que puede suspenderse cuando el paracetamol sea indetectable y se mantengan normales las cifras de GOT y GPT.
Si la paracetamolemia es negativa y las GOT/GPT son normales, la NAC es innecesaria. Se puede cursar el alta toxicológica.D. INTERVALO DESCONOCIDO Se procederá de forma idéntica a lo indicado en el supuesto C (intervalo > 24 h).

