










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
Print version ISSN 1137-6627
Anales Sis San Navarra vol.31 suppl.3 Pamplona 2008
Enfermedades sistémicas no infecciosas y uveítis
Non-infectious systemic diseases and uveitis
D. Díaz-Valle, R. Méndez, P. Arriola, R. Cuiña, M. Ariño
Unidad de Superficie e Inflamación Ocular. Hospital Clínico San Carlos. Madrid.
Dirección para correspondencia
RESUMEN
La uveítis se define como la inflamación del tracto uveal, aunque en la práctica clínica hace referencia a cualquier proceso inflamatorio intraocular. El origen de esta inflamación puede atribuirse a un mecanismo endógeno, ya sea formando parte de una enfermedad sistémica (sarcoidosis, enfermedad de Behçet, esclerosis múltiple, síndrome de Vogt-Koyanagi-Harada, etc.) o de forma ocular aislada. En muchas ocasiones, la enfermedad ocular constituye la forma de comienzo de una enfermedad sistémica. Por otra parte, la afectación ocular constituye, en ocasiones, la principal causa de morbilidad derivada de la enfermedad, por lo que su diagnóstico y tratamiento precoz son de la máxima importancia para evitar secuelas irreversibles. En este artículo se revisan los aspectos clínicos, diagnósticos y terapéuticos más relevantes de la afectación ocular en el contexto clínico de las enfermedades sistémicas no infecciosas más comúnmente asociadas a uveítis.
Palabras clave: Uveítis. Enfermedad de Behçet. Sarcoidosis. Esclerosis múltiple. Síndrome de Vogt-Koyanagi-Harada.
ABSTRACT
Uveitis can be defined as any inflammation affecting the uveal tract, although in clinical practice this term includes any intraocular inflammatory event. The etiology of this inflammation can be related to an endogenous mechanism in the clinical course of a systemic disease (sarcoidosis, Behçets disease, multiple sclerosis, Vogt-Koyanagi-Harada disease, etc.), or an isolated ocular entity. Sometimes, ocular inflammation is the initial manifestation of an undiagnosed systemic disease. On the other hand, ocular involvement could be the main cause of morbidity of the disease, and early diagnosis and treatment is an important issue in order to avoid irreversible ocular damage. In this article, the authors review some relevant clinical, diagnostic and therapeutic topics related to the most common non-infectious systemic diseases associated with uveitis.
Key words: Uveitis. Behçet disease. Sarcoidosis. Multiple sclerosis. Vogt-Koyanagi-Harada syndrome.
Introducción
En sentido estricto la uveítis se define como inflamación del tracto uveal. En la práctica clínica, este término se emplea para describir las enfermedades que afectan tanto al tracto uveal (iris, cuerpo ciliar y coroides) como a las estructuras adyacentes (vítreo, retina, nervio óptico y vasos). En definitiva, el término genérico uveítis hace referencia a cualquier proceso inflamatorio intraocular.
El origen de esta inflamación puede atribuirse a un mecanismo endógeno, ya sea formando parte de una enfermedad sistémica o de forma ocular aislada (síndromes primariamente oculares o «uveítis oftalmológicas»), o bien a un mecanismo exógeno, sobre todo en relación con agentes infecciosos que afecten el globo ocular de forma aislada o en el contexto de una infección multisistémica.
La uveítis representa un verdadero problema de salud, puesto que supone la tercera causa de ceguera en edades medias de la vida en países desarrollados; y se sabe que un tercio de los pacientes con uveítis atendidos en centros de referencia desarrollan pérdida visual (20/60) al menos en un ojo. Por otra parte, es importante conocer los diferentes síndromes uveíticos y sus características particulares puesto que con frecuencia se asocian a diferentes enfermedades sistémicas infecciosas y no infecciosas, y en muchos casos la uveítis constituye el debut de la enfermedad.
En esta revisión, se presentan los aspectos más relevantes de diferentes cuadros de uveítis que aparecen en el contexto de enfermedades sistémicas no infecciosas: sarcoidosis, enfermedad de Vogt-Koyanagi-Harada, esclerosis múltiple, enfermedad de Behçet, y otras enfermedades cuya asociación con cuadros de uveítis es menos frecuente como el lupus eritematoso sistémico (LES), panarteritis nodosa (PAN) y granulomatosis de Wegener.
Sarcoidosis
La sarcoidosis es una enfermedad granulomatosa multisistémica de etiología desconocida que puede afectar cualquier órgano de la economía, especialmente los pulmones, los ganglios linfáticos torácicos, la piel y los ojos. El diagnóstico definitivo se basa en la demostración histopatológica de granulomas epitelioides no caseificantes en los tejidos afectos. La enfermedad afecta más frecuentemente a adultos entre 20-40 años y es más prevalente en mujeres1.
Con frecuencia, el curso es agudo o subagudo y desaparece espontáneamente, pero en algunos individuos es crónico y persistente durante años. En estos casos, la incidencia de afectación ocular es más elevada2.
Manifestaciones clínicas
El proceso puede ser asintomático y descubrirse de forma casual en una radiografía de tórax rutinaria como una adenopatía hiliar o una infiltración del parénquima pulmonar; o bien iniciarse de forma aguda o subaguda y aparecer en un periodo de semanas o meses un cuadro de fiebre, mal estado general y tos seca, que puede acompañarse de uveítis, artritis y eritema nodoso. Los hallazgos típicos, en cualquier caso, son las adenopatías hiliares y paratraqueales (Fig. 1a).

En la forma de presentación aguda se han descrito dos síndromes. El síndrome de Löfgren, que se caracteriza por la presentación de eritema nodoso (Fig. 1b) y lesión mediastínica, y el síndrome de Heerfordt, que cursa con fiebre, parotiditis, uveítis y parálisis facial.
Otra forma de presentación es la insidiosa que se desarrolla durante meses, y suele consistir en síntomas respiratorios sin síntomas generales. Estos pacientes suelen desarrollar sarcoidosis crónica con lesiones infiltrativas, sobre todo a nivel pulmonar, cutáneo, articular y ocular. El pulmón es el órgano más frecuentemente afectado, de forma que un 90% de los casos presentan una radiografía de tórax anormal en algún momento de la evolución, y es la principal causa de morbilidad de esta enfermedad. La afectación ocular, por su parte, es la complicación extrapulmonar más frecuente3.
Sarcoidosis ocular
La incidencia de afectación ocular y, en su caso, el patrón de afectación es muy variable en el caso de la sarcoidosis. En general, se estima que un 20-50% de las sarcoidosis presentan lesiones oculares. Es importante destacar que puede adoptar cualquier patrón de uveítis, y que, por tanto, al igual que la sífilis y la tuberculosis debe descartarse siempre. Además, la inflamación puede localizarse a cualquier nivel del globo ocular, desde la conjuntiva hasta el nervio óptico4.
Aunque no existe un patrón ocular específico, determinadas formas clínicas oculares como la uveítis anterior granulomatosa, la uveítis intermedia bilateral, la panuveítis bilateral o las lesiones granulomatosas a nivel coroideo o de nervio óptico se consideran indicativas de sarcoidosis en presencia de un cuadro radiológico compatible5.
La uveítis anterior granulomatosa se considera la forma más frecuente de afectación ocular (53-60% de los pacientes). Se caracteriza por la presencia de precipitados retroqueráticos gruesos en grasa de carnero (Fig. 1c), marcado flare y células en cámara anterior, nódulos granulomatosos en el iris a nivel pupilar (nódulos de Koeppe) o sobre el estroma iridiano (de Bussaca), así como sinequias posteriores densas. Si no se instaura un tratamiento adecuado, pueden desarrollarse múltiples secuelas oculares como catarata, glaucoma y queratopatía en banda.
Los pacientes con uveítis intermedia suelen presentar vitritis, agregados celulares en el vítreo inferior (snowballs), exudados sobre la pars plana, infiltrados retinianos, vasculitis retiniana periférica, en ocasiones con los típicos exudados perivasculares en gota de cera (Fig.1 d), así como otras complicaciones con mayor implicación visual como el edema macular quístico.
Las lesiones coroideas pueden ser multifocales y localizarse en cualquier lugar del fondo de ojo, sin embargo, predominan en la hemirretina inferior, y suelen adoptar un aspecto en sacabocado. Este cuadro, que es más típico de mujeres de raza blanca, entre 30-50 años, debe hacer investigar la existencia de sarcoidosis6. Otra foma de afectación es el granuloma coroideo aislado.
Diagnóstico
En un caso típico, el diagnóstico de sarcoidosis puede establecerse basándose en una combinación de datos clínicos, radiológicos e histológicos. Sin embargo, a menudo los hallazgos son más sutiles.
Ningún dato de laboratorio es diagnóstico de la enfermedad, pero entre los hallazgos más frecuentes destaca la hipercalcemia en un 10%, hipercalciuria y elevación de la VSG. El aumento de los niveles de enzima conversor de la angiotensina (ECA) no es específico, puesto que puede elevarse en otras enfermedades. Además, en las formas de la enfermedad confinadas al ojo, la ECA no siempre aumenta puesto que es escasa la cantidad de tejido granulomatoso en el organismo. La radiografía de tórax, el TAC torácico y la gammagrafía con galio, junto con la determinación de ECA, constituye el método diagnóstico no invasivo más sensible para el diagnóstico de sarcoidosis7.
Sea clásica o no la forma de presentación, para establecer el diagnóstico definitivo se requiere la demostración histopatológica de los típicos granulomas no caseificantes en las muestras obtenidas de los tejidos afectos.
En cuanto a la sarcoidosis ocular, debe monitorizarse la afectación del segmento posterior con pruebas como la angiografía fluoresceínica (AFG) y especialmente con la angiografía con verde indocianina (AVI), puesto que según algunos autores, la participación coroidea ocurre en todos los casos con afectación posterior, pero no todos tendrán alteraciones retinianas. El signo más frecuentemente encontrado es la presencia de áreas difusas de hiperfluorescencia en fases tardías de la AVI debido a la tinción coroidea.
El diagnóstico diferencial es amplio y complicado en ocasiones, y debe hacerse con otras enfermedades granulomatosas como la tuberculosis, pero también con otros cuadros como la toxoplasmosis, la sífilis, el herpes o el linfoma intraocular.
Tratamiento
Considerando que algunos casos son oligosintomáticos y remiten sin tratamiento, en ocasiones es difícil decidir qué pacientes deben recibir tratamiento. En líneas generales, se considera que la afectación pulmonar progresiva, la afectación ocular y la enfermedad cardiaca deben ser tratadas. La base del tratamiento son los glucocorticoides, a dosis de 1 mg/kg/día de prednisona durante 4-6 semanas hasta controlar la inflamación, seguido por un descenso gradual durante 2-3 meses. Este ciclo se repetirá si la enfermedad recupera de nuevo su actividad. El metotrexato puede ser útil en los pacientes no respondedores a corticoides o para disminuir su dosis, evitando así los efectos adversos derivados del tratamiento corticoideo crónico8.
En la sarcoidosis ocular, los corticoides son también los fármacos de primera línea, administrados por vía tópica o periocular en función del patrón de afectación. Las indicaciones para el uso sistémico de corticoides en la uveítis sarcoidea son la presencia de uveítis posterior, neuritis óptica o uveítis intermedia bilateral refractaria al tratamiento periocular.
Al igual que en la sarcoidosis sistémica, los fármacos inmunosupresores y las nuevas terapias biológicas, especialmente infliximab, se emplean en casos refractarios, corticodependientes y/o con amenaza de la función visual, especialmente cuando existe una afectación sistémica importante9.
El pronóstico de las lesiones oculares es bueno si el tratamiento se inicia de forma precoz e intensiva.
Enfermedad de Vogt-Koyanagi-Harada
Vogt en 190610 y Koyanagi en 192911 describieron la coexistencia de uveítis anterior bilateral, vitíligo, poliosis, alopecia y disacusia. En 1926 Harada presentó un paciente con uveítis posterior, desprendimiento de retina y pleocitosis del líquido cefalorraquídeo12. Posteriormente se observó que dichos hallazgos formaban parte de una sola entidad, la enfermedad de Vogt-Koyanagi-Harada (VKH) o síndrome uveomeningítico, que se puede definir como una patología inflamatoria granulomatosa, multisistémica, de etiología desconocida, caracterizada por una respuesta autoinmune, mediada por células T, dirigida contra antígenos melanocíticos del ojo, piel, sistema nervioso y auditivo. La manifestación característica de la enfermedad a nivel ocular es en forma de panuveítis bilateral severa asociada a desprendimiento de retina exudativo.
Diagnóstico
Diferentes criterios han sido utilizados para el diagnóstico y clasificación de la enfermedad de VKH, actualmente los más aceptados son los criterios revisados en 2001 sobre los propuestos en 1999 por el Primer Grupo Internacional de Trabajo de la enfermedad de VKH (First VKH International Workshop Group)13, diferenciando entre VKH completo (afectación ocular + neurológica/auditiva + dermatológica), VKH incompleto (afectación ocular + neurológica/auditiva o dermatológica) y VKH probable (sólo afectación ocular).
Clínica
Las manifestaciones clínicas varían dependiendo de la fase evolutiva14.
– Fase prodrómica: síntomas gripales inespecíficos, síntomas neurológicos (cefalea o meningismo, raramente signos focales neurológicos), hasta un 70% de los pacientes presenta hiperalgesia del cuero cabelludo.
– Fase uveítica aguda: disminución de la agudeza visual bilateral por afectación del polo posterior con desprendimiento seroso de retina (Fig 2), hiperemia-edema del nervio óptico y engrosamiento coroideo. Puede existir uveítis anterior.

– Fase crónica: despigmentación en piel y/o úvea (vitíligo, sunset glow fundus, focos de hipo y/o hiperpigmentación, áreas redondeadas de atrofia coriorretiniana en media periferia, signo de Sugiura).
– Fase crónica recurrente: en algunos pacientes, debido al curso de la enfermedad o por un tratamiento insuficiente, la inflamación se hace crónica y recurrente, generalmente en forma de uveítis anterior, aunque puede existir afectación posterior asociada.
Pruebas diagnósticas
Aunque el diagnóstico es fundamentalmente clínico, existen pruebas complementarias de apoyo.
– Angiografía fluoresceínica: zonas de retraso del llenado coroideo en tiempos precoces, seguido por múltiples puntos de fuga, que pueden coalescer en imágenes placoides, rellenándose en tiempos tardíos los desprendimientos serosos (Fig 1).
– Angiografía con verde de indocianina: puede detectar recurrencias subclínicas15.
– Punción lumbar (indicada en casos dudosos): pleocitosis linfocítica, pudiéndose identificar macrófagos cargados de melanina.
– Tomografía de coherencia óptica (OCT): para identificar y realizar el seguimiento de los desprendimientos serosos.
Tratamiento
Debe iniciarse precozmente ya que cuanto antes se logre el control de la inflamación menores serán las secuelas y mejor el resultado final16-17.
Corticoides
Los corticoides sistémicos son el tratamiento de elección del VKH. Se recomienda el uso de pulsos intravenosos de hasta 1 g de metilprednisolona/día durante tres días consecutivos, continuando con 1 mg/Kg/día de prednisona vía oral18. En la mayoría de los casos, un tratamiento precoz y agresivo con corticoides sistémicos va a suprimir el proceso inflamatorio agudo y minimizar el desarrollo de complicaciones. El tratamiento debe mantenerse a dosis altas hasta conseguir una clara mejoría, habitualmente no menos de 4 semanas y después seguir una pauta de descenso lenta en 6-9 meses (nunca menos de 3-6 meses)19-20 para así disminuir las posibilidades de una recurrencia rápida y mejorar el pronóstico visual21. En determinados casos, como refuerzo de la terapia sistémica, pueden ser útiles los corticoides subtenonianos22 y/o intravítreos23.
Inmunosupresores y terapias biológicas
Puede ser necesario asociar inmunosupresores al tratamiento esteroideo en casos de control insuficiente o intolerancia corticoidea. Como inmunosupresores se han utilizado sobre todo la ciclosporina, la azatioprina y el metotrexato, solos o combinados entre sí24. Algunos autores utilizan los inmunosupresores como tratamiento de primera línea25.
Otros tratamientos utilizados en esta patología con buenos resultados son los anticuerpos monoclonales contra el Factor de Necrosis Tumoral alfa (anti-TNFa), entre ellos el infliximab26 y el adalimumab27.
Esclerosis múltiple y uveítis
La esclerosis múltiple (EM) es una enfermedad crónica desmielinizante y neurodegenerativa del sistema nervioso central. Existen distintas formas de evolución de la enfermedad siendo la más frecuente la remitente-recurrente (EMRR) que afecta a más del 80% de las personas con EM Su fisiopatología es compleja y están involucrados factores genéticos y medioambientales junto con el desarrollo de una respuesta autoinmune patológica que induce la destrucción del la mielina, la pérdida axonal y la presencia de infiltrados inflamatorios locales28. La enfermedad suele debutar en mujeres de entre 20 y 50 años, pero puede aparecer en edades más avanzadas y hasta en un 5% afecta a menores de 16 años29.
La afectación ocular más frecuente es la neuritis óptica que aparece hasta en un 50% de los pacientes con EM y en un 20% puede ser la manifestación inicial de la enfermedad30-31. Entre otras manifestaciones oculares asociadas a la EM aparecen las alteraciones motoras (parálisis de los pares craneales, nistagmus, etc ) y las inflamaciones oculares (vasculitis retinianas y uveítis).
Las vasculitis retinianas se asocian a la EM en un 10-39% de pacientes. Se caracterizan por una exudación perivascular, hemorragias y oclusiones venosas. Las complicaciones son poco frecuentes, pero graves (neovascularización retiniana, hemorragias vítreas, desprendimientos de retina traccionales )32-33. Se ha encontrado una importante correlación entre la presencia de vasos envainados y la progresión de la discapacidad. La presencia de periflebitis es, por tanto, un indicador de la actividad sistémica de la enfermedad.
Las uveítis constituyen una manifestación poco frecuente de la EM. Su incidencia se estima alrededor de un 1%34. Afectan, típicamente, a mujeres de entre 20-50 años y caucásicas. Preceden a la enfermedad en un 25% de pacientes y aparecen de forma concomitante en un 19% de éstos35. El 94% son bilaterales. La gran mayoría de publicaciones coinciden en que las formas más frecuentes son la pars planitis (uveítis intermedia) y las panuveítis36. La incidencia de EM en pacientes con uveítis intermedias es de un 16%. Un gran número de estos pacientes son HLA DR15 +. Otros autores, sin embargo, describen las uveítis anteriores granulomatosas y bilaterales como el tipo de uveítis más frecuentemente asociado a la EM. Ante este tipo de uveítis anteriores se debe hacer el diagnóstico diferencial con otras entidades como el síndrome de Vogt-Koyanagi-Harada, la sarcoidosis, la tuberculosis y la sífilis.
El uso de terapias biológicas en el tratamiento de las uveítis está cada vez más extendido. Conocemos el claro potencial de los anti-TNF para exacerbar y acelerar las enfermedades desmielinizantes9. Es, por tanto, necesario descartar la existencia de una EM antes de iniciar el tratamiento ya que su diagnóstico contraindica el uso de estos fármacos en las uveítis.
No ha quedado demostrada, hasta ahora, una relación clínico-pronóstica entre la presencia de uveítis y las distintas formas de evolución de la EM.
Enfermedad de Behçet
La enfermedad de Behçet (EB) es una enfermedad inflamatoria crónica, de curso episódico, y de etiología desconocida.
Descrita por el dermatólogo turco H. Behçet en 1937, originalmente la enfermedad fue definida con la triada de úlceras orales y genitales recurrentes, además de una uveítis con hipopion. Con los años, el espectro de la enfermedad se ha ampliado con múltiples manifestaciones (enfermedad multisistémica).
No existe ningún dato clínico ni de laboratorio que sea patognomónico de la EB. El diagnóstico se ha basado en criterios clínicos. Los más ampliamente usados desde 1990, son los criterios del Grupo de Estudio Internacional para la Enfermedad de Behçet (Tabla 1)37. En 2003, el Comité de Investigación Japonés de la EB hizo una revisión de estos criterios (Tabla 2)38.

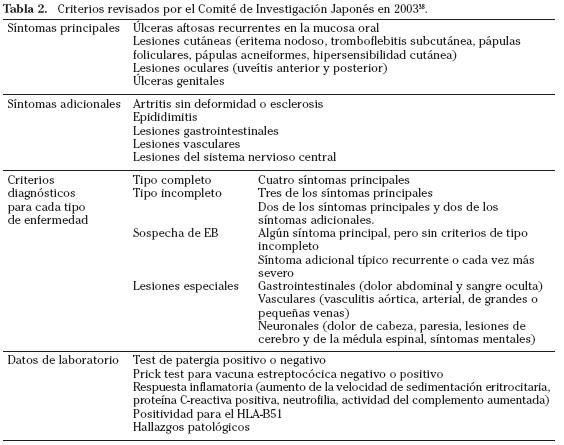
La distribución de la EB se localiza predominantemente a lo largo de la antigua ruta de la seda, desde los países mediterráneos hasta oriente medio y este asiático. La mayor prevalencia se ha notificado en Turquía, y más concretamente en Anatolia (370 por 100.000 habitantes). En España es baja, aproximadamente 5 de cada 100.000 habitantes. La prevalencia y expresión de la enfermedad estaría determinada tanto por factores genéticos como ambientales39-40.
El origen de la enfermedad es desconocido, aunque se han sugerido teorías como la vírica, la inmunológica y la genética para explicar su patogenia. Se considera un síndrome multisistémico cuya lesión básica es una vasculitis de pequeños vasos, con una infiltración perivascular linfo-monocitaria y una respuesta quimiotáctica exagerada de los polimorfonucleares. Existe asociación del antígeno HLA-B5 hasta en un 30-40% de los pacientes con EB en Japón y otras zonas mediterráneas. También algunos autores han sugerido el posible papel de un agente viral en la etiología de la enfermedad41.
Clínica
Manifestaciones extraoculares
La manifestación clínica más frecuente de la enfermedad de Behçet son las aftas bucales recidivantes. También son típicas las aftas genitales, localizadas en el hombre en el escroto y el pene, y en la mujer en la vulva y la mucosa genital. Las lesiones cutáneas pueden ser diversas, siendo la más frecuente la similar al eritema nodoso en extremidades inferiores, aunque también pueden aparecer vesículas, pústulas, foliculitis, y otras.
Manifestaciones oculares
La afectación ocular aparece en el 70% de los pacientes con EB, y supone un alto riesgo de ceguera42. La causa más frecuente de pérdida de visión en los pacientes con EB es la turbidez inflamatoria, seguida de las cataratas secundarias y el edema macular quístico43. Generalmente, la inflamación se localiza en el segmento posterior del ojo, aunque en ocasiones puede asociarse a una uveítis anterior, típicamente con hipopion. Los brotes más típicos se caracterizan por la presencia de infiltrados retinianos blanquecinos acompañados de periflebitis y vitritis (Fig 3).

Tratamiento
La EB es una enfermedad multisistémica y precisa un enfoque interdisciplinario. Debido al riesgo de pérdida importante de visión, se debe aplicar un tratamiento precoz y correcto de la afectación ocular. Así, la medicación administrada debería ser eficaz para controlar la inflamación ocular y disminuir la frecuencia e intensidad de los brotes. También debe ser útil para tratar la afectación extraocular de la EB.
La respuesta al tratamiento está influida por la edad de los pacientes y el tipo de lesión retiniana. En los pacientes jóvenes la afectación ocular suele ser de curso más explosivo y evolución más agresiva, y son más frecuentes las lesiones como vasculitis, infiltrados retinianos y vitritis severa, que pueden desarrollar secuelas irreversibles sobre la función visual.
Las pautas de tratamiento incluyen:
1. Tratamiento tópico con colirios de corticoides y midriáticos para la uveítis anterior. El tratamiento corticoideo puede administrarse por vía periocular en casos refractarios o en los que desarrollan complicaciones como el edema macular quístico.
2. Tratamiento sistémico: consideramos diferentes niveles de tratamiento:
– Primer escalón. Combinación de corticoide y ciclosporina. Se debe comenzar el tratamiento con prednisona oral (1 mg/kg/día). Generalmente con esta dosis se controla la inflamación si es un primer episodio, pero para mantener el control de la enfermedad a largo plazo es preciso asociar ciclosporina A (5 mg/kg/día), que nos permitirá reducir los esteroides. Por este motivo, en la enfermedad de Behçet con afectación del segmento intermedio o posterior se comienza con la terapia combinada de corticoides y ciclosporina a las dosis indicadas para posteriormente reducir la dosis de corticoides hasta la dosis mínima eficaz y continuar con ciclosporina como tratamiento de mantenimiento.
– Segundo escalón. En casos refractarios, que desarrollan efectos adversos inaceptables o que recidivan al reducir la dosis de corticoide con una pauta adecuada, se asocia un segundo inmunosupresor como metotrexato, azatioprina o micofenolato de mofetilo.
– Tercer escalón. Si a pesar del tratamiento indicado no se alcanza un control adecuado de la actividad inflamatoria, se emplean las terapias biológicas, como infliximab, adalimumab, etanercept o interferon44-45. Estos tratamientos han mostrado una excelente eficacia terapéutica antiinflamatoria en los casos en los que el patrón de afectación ocular predominante es vitritis, vasculitis y/o edema macular.
– Cuarto escalón. En el último escalón terapéutico se sitúan los agentes alquilantes como clorambucilo o ciclofosfamida, cuyo empleo en la actualidad se reserva a casos muy seleccionados con mala respuesta a los tratamientos indicados, puesto que estos fármacos se asocian a importantes efectos adversos46.
En algunos casos puede ser necesario la administración de corticoides por vía intravítrea o los nuevos fármacos anti-VEGF (factor de crecimiento endotelial vascular) para el control de algunas secuelas inflamatorias como el edema macular refractario al tratamiento convencional o la neovascularización subretiniana47.
Lupus eritematoso sistémico (LES)
El LES presenta manifestaciones sistémicas además de las cutáneas. Dentro de las manifestaciones oculares, el hallazgo más frecuente es la existencia de una vasculitis retiniana48, en cuya patogénesis, más que un auténtico fenómeno inflamatorio, se ha implicado la presencia de anticuerpos antifosfolípido, entre ellos los anticuerpos anticardiolipina y anticoagulante lúpico, que confieren a estos pacientes un riesgo aumentado de fenómenos trombóticos sistémicos. También parece participar en su etiología el depósito de complejos inmunes. La prevalencia de una vasculitis clínicamente detectable se ha estimado en torno al 10% de los casos con LES, aumentando dicha cifra al 29% empleando angiofluoresceingrafía para el diagnóstico, pues muchos de los casos son asintomáticos49 (Fig. 4). La vasculitis retiniana se manifiesta como una enfermedad vasooclusiva grave, con exudados algodonosos (probablemente el signo más característico de la afectación ocular por LES) secundarios a vasculitis oclusivas de pequeños vasos arteriales, así como hemorragias intrarretinianas, exudados duros, microaneurismas y alteraciones vasculares (afilamiento arteriolar, arrosariamiento venoso y tortuosidad vascular). La vasculitis puede resultar en una retinopatía proliferativa secundaria a la isquemia retiniana, apareciendo neovascularización retiniana y hemorragias vítreas. La presencia de una vasculitis retiniana en el contexto del LES se asocia a la presencia de anticuerpos anticardiolipina, neurolupus y guarda relación con la gravedad de la enfermedad.

Mucho más infrecuente es la coroidopatía lúpica, caracterizada por la presencia de desprendimientos serosos de retina multifocales asociados a zonas parcheadas o moteadas de epitelio pigmentario de la retina (EPR). En su patogenia se ha implicado la presencia de alteraciones vasculares coroideas que provocan isquemia del EPR con trasudación, identificables mediante angiografía con verde de indocianina (ICG)50.
Se han descrito casos de uveítis anterior, aunque son excepcionales.
Poliarteritis Nodosa (PAN)
Se ha estimado que el 20% de los pacientes afectos de PAN presentan afectación ocular51, siendo la queratitis ulcerativa periférica la manifestación ocular más frecuente; la presencia de una vasculitis retiniana multifocal es también usual. Esta vasculitis afecta principalmente a las arteriolas y se deben a depósitos de complejos inmunes en la pared de los vasos. También se ha descrito la presencia de la llamada coroidopatía multifocal isquémica, que no es más que una vasculitis coroidea multifocal y bilateral, con manchas amarillentas en el fondo de ojo que pueden simular una epiteliopatía pigmentaria placoide posterior aguda o una coroiditis serpiginosa. También pueden aparecer uveítis anteriores fibrinosas aunque no es una presentación habitual.
Granulomatosis de Wegener (GW)
La afectación ocular ocurre entre un 28% y un 87% de los pacientes con GW, según las series52. Las manifestaciones oftalmológicas son el signo de presentación inicial de la enfermedad en el 16% de los casos. Al igual que en la PAN, la queratitis ulcerativa periférica es la manifestación ocular más frecuente. La uveítis anterior granulomatosa es la tercera en frecuencia (10-20%)53. También se han descrito otras formas de uveítis, como uveítis intermedia, vasculitis retiniana, coriorretinitis inflamatoria, necrosis retiniana aguda, etc54. Es importante recordar la elevada especificidad de los anticuerpos anticitoplasma de los neutrófilos, en concreto del patrón citoplásmico (cANCA), para su diagnóstico51.
Bibliografía
1. Fernandez Espartero MC, Alejandre N, Diaz-Valle D. Sarcoidosis. En Diaz-Valle D, Méndez R, Benitez del Castillo JM. Actualización en el tratamiento de las uveítis. Ed. Sociedad Española de Oftalmología. Madrid 2007; 123-130. [ Links ]
2. Crystal R. Sarcoidosis. En Harrison. Principios de Medicina Interna. Ed. Mc Graw-Hill: 1994; 1932-1938. [ Links ]
3. Maña M. Sarcoidosis. Med Clin (Barc) 2001; 116: 307-311. [ Links ]
4. Jabs DA, Johns CJ. Ocular involvement in chronic sarcoidosis. Am J Ophthalmol 1986; 102: 297-301. [ Links ]
5. Adán A, Baget M, Llobet JM, Segura A, Marieges MT, Casaroli-Marano R. Uveítis como manifestación inicial de sarcoidosis. Estudio de 31 pacientes. Med Clin (Barc) 2004; 122: 748-752. [ Links ]
6. Abad S, Meyssonier V, Allali J, Gouya H, Giraudet AL, Monnet D et al. Association of peripheral multifocal choroiditis with sarcoidosis: a study of thirty-seven patients. Arthritis Rheum 2004; 51: 974-982. [ Links ]
7. Power WJ, Neves RA, Rodríguez A, Pedroza-Seres M, Foster CS. The value of combined serum angiotensina converting enzyme and gallium scan in the diagnosis of sarcoidosis. Ophthalmology 1995; 102: 2007-2012. [ Links ]
8. Dev S, McCaluum RM, Jaffe GJ. Methotrexate treatment for sarcoid-associated panuveitis. Ophthalmology 1999 106: 111-118. [ Links ]
9. Pritchard C, Nadarajah K. Tumour necrosis factor-alpha inhibitor treatment for sarcoidosis refractory to convenctional treatments. A reporto f five patients. Ann Rheum Dis 2004; 63: 318-320. [ Links ]
10. Vogt A. Fruhzeitiges Ergaruen der Zilien und Bemerkungen uber den sogenaten plotzlichen Eintreitt dieser Veranderung. Klin Monatsbl Augenheilkd 1906; 44: 228-242. [ Links ]
11. Koyanagi Y. Dysakusis, alopecia und poliosis bei schwerer Uveitis nicht traumatischen Ursprugs. Klin Monatsbl Augenheilkd 1929; 82: 194-211. [ Links ]
12. Harada E. Acute diffuse choroiditis. Acta Soc Ophthalmol Jpn 1926; 30: 356-378. [ Links ]
13. Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001; 131: 647-652. [ Links ]
14. Andreoli CM, Foster CS. Vogt-Koyanagi-Harada disease. Int Ophthalmol Clin 2006; 46: 111-122. [ Links ]
15. Bouchenaki N, Herbort CP. The contribution of indocyanine green angiography to the appraisal and management of Vogt-Koyanagi-Harada disease. Ophthalmology 2001; 108: 54-64. [ Links ]
16. Bouchenaki N, Morisod L, Herbort CP. Vogt-Koyanagi-Harada syndrome: importance of rapid diagnosis and therapeutic intervention. Klin Monatsbl Augenheilkd 2000; 216: 290-294. [ Links ]
17. Bykhovskaya I, Thorne JE, Kempen JH, Dunn JP, Jabs DA. Vogt-Koyanagi-Harada disease: Clinical Outcomes. Am J Ophthalmol 2005; 140: 674-678. [ Links ]
18. Yamanaka E, Ohguro N, Yamamoto S, Nakagawa Y, Imoto Y, Tano Y. Evaluation of pulse corticosteroid therapy for Vogt-Koyanagi-Harada disease assessed by optical coherence tomography. Am J Ophthalmol 2002; 134: 454-456. [ Links ]
19. Lai TY, Chan RP, Chan CK, Lam DS. Effects of the duration of initial oral corticosteroid treatment on the recurrence of inlamation in Vogt-Koyanagi-Harada disease. Eye 2008, Mar 28. [Epub ahead of print] [ Links ]
20. Sasamoto Y, Ohno S, Matsuda H. Studies on corticosteroid therapy in Vogt-Koyanagi-Harada disease. Ophthalmologica 1990; 201: 162-167. [ Links ]
21. Rubsamen PE, Gass JD: Vogt-Koyanagi-Harada syndrome. Clinical course, therapy and long-term visual outcome. Arch Ophthalmol 1991; 109: 682-687. [ Links ]
22. Rajendram R, Evans M, Rao NA. Vogt-Koyanagi-Harada disease. Int Ophthalmol Clin 2005; 45: 115-134. [ Links ]
23. Andrade RE, Muccioli C, Farah ME, Nussenblatt RB, Belfort R Jr. Intravitreal triamcinolone in the treatment of serous retinal detachment in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol 2004; 137: 572-574. [ Links ]
24. Kim SJ, Yu HG. The use of low-dose azathioprine in patients with Vogt-Koyanagi-Harada disease. Ocul Immunol Inflamm 2007; 15: 381-387. [ Links ]
25. Paredes I, Ahmed M, Foster CS. Immunomodulatory therapy for VKH patients as first line therapy. Ocul Immunol Inflamm 2006; 14: 87-90. [ Links ]
26. Kahn P, Weiss M, Imundo LF, Levy DM. Favorable response to high-dose infliximab for refractory childhood uveitis. Ophthalmology 2006; 113: 860-864. [ Links ]
27. Díaz Llopis M, Amselem L, Romero FJ, García-Delpech S, Hernández ML. Adalimumab therapy for Vogt-Koyanagi-Harada syndrome. Arch Soc Esp Oftalmol 2007; 82: 131-132. [ Links ]
28. Hafler DA, Slavik JM, Anderson DE, Oconnor KC, De Jager P, Beacher-Allan C. Multiple sclerosis. Immunol Rev 2005; 204: 208-231. [ Links ]
29. Ruggieri M, Iannetti P, Polizzi A, Pavone L, Grimaldi LM, Italian Society of Paediatric Neurology Study Group on Childhood Multiple Sclerosis. Multiple sclerosis in children under 10 years of age. Neurol Sci 2004; 25 (Suppl 4): S326-S335. [ Links ]
30. Jacobs DA, Galetta SL. Multiple sclerosis and the visual system. Ophthalmol Clin North Am 2004; 17: 265-273. [ Links ]
31. Frohman EM, Frohman TC, Zee DS, et al. The neuro-ophthalmology of multiple sclerosis. Lancet Neurol 2005; 4: 111-121. [ Links ]
32. Towler HM, Lightman S. Symptomatic intraocular inflammation in multiple sclerosis. Clin Experiment Ophthalmol 2000; 28: 97-102. [ Links ]
33. Patte M, Rouher FN, Vernay D, Delaire JC, Bacin F. Proliferative retinal vasculitis and multiple sclerosis: a case report. J Fr Ophtalmol 2003; 26: 381-385. [ Links ]
34. Smith JR, Rosenbaum JT. Neurological concomitants of uveitis. Br J Ophthalmol 2004; 88: 1498-1499. [ Links ]
35. Zein G, Berta A, Foster CS. Multiple sclerosis-associated uveitis. Ocul Immunol Inflamm 2004; 12: 137-142. [ Links ]
36. Biousse V, Trichet C, Bloch-Michel E, Roullet E. Multiple sclerosis associated with uveitis in two large clinic-based series. Neurology 1999; 52: 179-181. [ Links ]
37. International Study Group for Behçets disease. Criteria for diagnosis of Behçet disease. Lancet 1990; 335: 1078-1080. [ Links ]
38. Suzuki Kurokawa M, Suzuki N. Behcets disease. Clin Exp Med. 2004; 4: 10-20. [ Links ]
39. Yazici H, Tuzun Y, Pazarli H, Yurdakul S, Ozyazgan, Ozdogan H et al. Influence of age of onset and patients sex on prevalence and severity of manifestations of Behcets syndrome, Ann Rheum Dis 1984; 43 : 783-789. [ Links ]
40. Díaz Llopis M, Salom Alonso D et al. Enfermedad de Behcet. Díaz-Valle D, Méndez Fernández R, Benitez del Castillo JM. Actualización en el tratamiento de las uveítis. Madrid. Sociedad Española de Oftalmología. 2007: 113-122. [ Links ]
41. Zafirakis P: Adamantiades-Behçet Disease. En: Foster CS, Vitale AT. Diagnosis and Treatment of Uveitis. Philadelphia, PA: W.B. Saunders Co; 2002; 632-652. [ Links ]
42. Verity DH, Wallace GR, Vaughan RW, Stanford MR. Behçets disease: from Hippocrates to the third millennium, Br J Ophthalmol 2003; 87: 1175-1183. [ Links ]
43. Kaçmaz RO, Kempen JH, Newcomb C, Gangaputra S, Daniel E, Levy-Clarke Ga et al. Ocular inflammation in Behçet disease: incidence of ocular complications and of loss of visual acuity. Am J Ophthalmol 2008 Aug. 15 [Epud ahead of print]. [ Links ]
44. Ohno S, Nakamura S. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behcet´s disease with refractory uveoretinitis. J Rheumatol 2004; 31: 1362-1368. [ Links ]
45. Sfikakis PP, Kaklamanis PH. Infliximab for recurrent, sightthreatening ocular inflammation in Adamantiades-Behcetdisease. Ann Intern Med 2004; 140: 404-406. [ Links ]
46. Tessler HH, Jennings T. High-dose short-term chlorambucil for intractable sympathetic ophthalmia and Behcets disease. Br J Ophthalmol 1990; 74: 353-357. [ Links ]
47. Adán A, Navarro M, Casaroli-Marano RP, Ortiz S, Molina JJ. Intravitreal bevacizumab as initial treatment for choroidal neovascularization associated with presumed ocular histoplasmosis syndrome. Graefes Arch Clin Exp Ophthalmol 2007; 245: 1873-1875. [ Links ]
48. Mondéjar JJ, Díaz-Llopis M. Lupus Eritematoso. En: Sánchez Salorio M, Díaz-Llopis M, Benítez del Castillo JM, Rodríguez Ares MT. Manifestaciones oftalmológicas de las enfermedades generales. Madrid; Sociedad Española de Oftalmología; 2001: 69-72. [ Links ]
49. Díaz-Valle D, Santos Bueso E, Mígueles Sanchez R, Díaz-Valle T. Vasculitis retiniana. En Díaz-Valle D, Méndez Fernández R, Benítez del Castillo JM. Actualización en el tratamiento de las uveítis. Madrid; Sociedad Española de Oftalmología; 2007: 141-148. [ Links ]
50. Gharbiya M, Bozzoni-Pantaleoni F, Augello F, Balacco-Gabrieli C. Indocyanine green angiographic findings in systemic lupus erythematosus choroidopathy. Am J Ophthalmol 2002; 134: 286-290. [ Links ]
51. Nussenblatt RB. Retinal Vasculitis. Nusenblatt RB, Whitcup SM. Uveitis. Philadelphia, Pennsylvania, USA; Mosby; 2004: 372-383. [ Links ]
52. Mondéjar JJ, Díaz-Llopis M. Granulomatosis de Wegener. En: Sánchez Salorio M, Díaz-Llopis M, Benítez del Castillo JM, Rodríguez Ares MT. Manifestaciones oftalmológicas de las enfermedades generales. Madrid; Sociedad Española de Oftalmología; 2001: 67-69. [ Links ]
53. Huong du LT, Tran TH, Piette JC. Granulomatous uveitis revealing Wegeners granulomatosis. J Rheumatol 2006; 33: 1209-1210. [ Links ]
54. Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA. Ocular complications of Wegeners granulomatosis. Ophthalmology 1983; 90: 279-290. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
David Díaz-Valle
Unidad de Superficie e Inflamación Ocular
Hospital Clínico San Carlos
Martín Lagos, s/n
28040 Madrid
Tfno. 913303963
Fax 913303975
E-mail: daviddiazvalle@yahoo.es

