










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.35 no.1 Pamplona ene./abr. 2012
https://dx.doi.org/10.4321/S1137-66272012000100011
Feocromocitoma. Informe de un caso
Pheochromocytoma. A case report
B. Rípodas1, A. Arillo1, M. Murie2 y D. García García3
1. Centro de Salud Chantrea. Pamplona
2. Servicio de Medicina Interna. Complejo Hospitalario de Navarra
3. Servicio de Urología. Complejo Hospitalario de Navarra
Dirección para correspondencia
RESUMEN
Se presenta el caso de un varón de 34 años de edad, a quien se diagnosticó un feocromocitoma maligno y fue tratado con suprarrenalectomía izquierda lapararoscópica. Se trata de una neoplasia de las células cromafines con una prevalencia de dos casos por millón de habitantes, que, generalmente, causa los síntomas típicos de liberación episódica de catecolaminas. Se describe la forma de presentación del caso, que debuta con episodios sucesivos de dolorimiento abdominal intenso en flanco izdo. En la ecografía abdominopélvica y TAC abdominal, se objetiva una masa de 6,5 cms de diámetro, dependiente de la glándula suprarrenal izda. Se evidencian niveles elevados de metanefrinas y catecolaminas en orina. Se realiza adrenalectomía total izquierda laparoscópica. La anatomía patológica evidencia feocromocitoma maligno.
Palabras clave: Feocromocitoma. Adrenalectomía. Catecolaminas. Paraganglioma. Malignidad.
ABSTRACT
We present the case of a 34 year-old male, who was diagnosed with a malign pheochromocytoma and who was treated with left laparascopic suprarenalectomy. This is a neoplasia of the chromoffin cells with a prevalence of two cases per million inhabitants, which generally causes the typical symptoms of episodic freeing of catecholamines. We describe the case´s form of presentation, which began with successive episodes of intense abdominal pain on the left side. In the abdominal pelvic ultrasound and abdominal CT, a mass of 6.5 cm diameter was objectified, dependent on the left suprarenal gland. High levels of metanefrines and catecholamines were evident in the urine. A total left laparoscopic adrenalectomy was carried out. The pathological anatomy showed malign pheochromocytoma.
Key words: Pheochromocytoma. Adrenalectomy. Catecholamines. Paraganglioma. Malignity.
Introducción
El feocromocitoma es un tumor productor de catecolaminas que procede de las células cromafines del sistema nervioso simpático1. Representa aproximadamente el 0,2% de los casos de hipertensión de causa curable2,3. El 80-85% se localiza en la médula adrenal y el 15-20% son de localización extraadrenal y se denominan paragangliomas. Pueden originarse en cualquier lugar donde exista tejido cromafín: a lo largo de la cadena ganglionar simpática paraaórtica, en el órgano de Zuckerkandl (en el origen de la arteria mesentérica inferior), en la pared de la vejiga urinaria y en la cadena ganglionar simpática en el cuello o el mediastino4. La distinción entre un feocromocitoma verdadero y un paraganglioma es importante debido al diferente compartamiento en cuanto al riesgo de malignidad, la posibilidad de otras neoplasias asociadas y la necesidad de estudios genéticos.
La incidencia de esta enfermedad es de dos casos por millón de habitantes1 y representa entre 0,3-1,9% de los causas secundarias de hipertensión arterial en la población general5. Afecta con igual frecuencia a ambos sexos, salvo en la población infantil, donde se encuentra con más frecuencia entre los varones (60%). La incidencia aumenta con la edad, que al diagnóstico oscila entre los 30-50 años. Los feocromocitomas suponen el 6,5% de los incindentalomas suprarrrenales6. Pueden ser esporádicos o encontrarse asociados a varias enfermedades genéticas como son la neoplasia endocrina múltiple 2, la enfermedad de Von Hippel-Lindau, la neurofibromatosis de tipo 1 y el paraganglioma familiar con mutaciones en la succinato deshidrogenasa.
Las manifestaciones clínicas son producto de la secreción excesiva de catecolaminas. Se le ha llamado "el gran imitador" ya que da síntomas muy variados dependiendo de la catecolamina en exceso. Aparecen en forma de crisis paroxísticas de 15-60 minutos de duración y resolución lenta, cuya frecuencia aumenta con el tiempo de evolución. La tétrada clásica consiste en cefalea (80%), palpitaciones (64%), diaforesis (57%) e hipertensión arterial (85%). La hipertensión puede ser mantenida en la mitad de los pacientes y paroxística en un tercio, mientras el resto de los sujetos presentan normotensión. Además de estos síntomas pueden aparecer dolor abdominal, vómitos, dolor torácico, taquicardia, nerviosismo, irritabilidad, pérdida de peso, temblor de manos, palidez, frialdad y humedad de las manos, aumento de la temperatura y flushing. Las crisis se siguen habitualmente de intensa fatiga.
Se hace alusión a la regla del 10: el 10% son extraadrenales; el 10% se presentan en niños; el 10% son múltiples o bilaterales; el 10% recidiva tras la cirugía; el 10% son malignos; el 10% son familiares; el 10% son descubiertos como incindentalomas adrenales. No obstante, estudios recientes han demostrado que hasta un 25% son familiares7.También se ha descrito la presencia de feocromocitoma hasta en un 57% de pacientes con incendentaloma adrenal8.
Se debe sospechar de un feocromocitoma en las siguientes situaciones: hipertensión resistente al tratamiento, crisis adrenérgicas, historia familiar de feocromocitoma, síndrome genético que predisponga, incidentaloma adrenal radiológicamente compatible, hipertensión en paciente joven y respuesta presora durante la inducción de la anestesia.
Es importante sospechar, confirmar, localizar y resecar el feocromocitoma por varias causas:
- Hipertensión arterial asociada, curable con la resección quirúrgica del tumor.
- Riesgo de muerte súbita.
- Presentar malignidad en un 10% de los casos.
- La detección en los casos de afección familiar puede resultar útil en el diagnóstico precoz de otros miembros de la familia.
El diagnóstico del feocromocitoma debe establecerse bioquímicamente mediante la determinación de metanefrinas plasmáticas y/o urinarias9. Para confirmar el diagnóstico, el resultado de las determinaciones hormonales debe ser por lo menos el doble del límite superior del rango de referencia. El diagnóstico de localización inicialmente debe basarse en la realización de tomografía computarizada (TC) o resonancia magnética (RM). Además deben realizarse sólo del abdomen inicialmente. Aproximadamente entre el 9 y el 23% de los tumores son extraadrenales10,11, pero el 95% se localiza en el abdomen y la pelvis12. La gammagrafía con 123I-metayodobencilguanidina es la prueba funcional de imagen de elección. La tomografía por emisión de positrones ha resultado de utilidad en enfermedad metastásica.
El tratamiento de elección es la cirugía por vía laparoscópica (la adrenalectomía debe ser completa), después de la realización del bloqueo alfaadrenérgico, en tumores menores de 8 cm, ya que ofrece varias ventajas respecto a la cirugía convencional13,14. No está indicada cuando hay evidencia preoperatoria de infiltración de los tejidos circundantes15,16.
Aproximadamente el 10% de los feocromocitomas son malignos y hasta un 50% de los paragangliomas son malignos. Histológicamente y bioquímicamente son similares. La única prueba evidente de la presencia de un feocromocitoma maligno es la invasión local o la presencia de metástasis y puede ocurrir hasta 20 años después de la resección de tumor17. El tratamiento de elección es la exéresis del tumor. Para la enfermedad inoperable puede utilizarse la quimioterapia.
El pronóstico es bueno, excepto en los casos de enfermedad maligna donde la tasa de supervivencia a los 5 años es menor del 50%. El conocimiento de nuevos genes causantes de enfermedad hereditaria ha supuesto un cambio en las recomendaciones sobre la necesidad de realizar estudio genético5.
Caso clínico
Varón de 34 años, fumador de 15 cigarros al día, con antecedentes de esofagitis péptica, hernia de hiato y asma bronquial no alérgico. Padre diabético. Cuadro de dolor abdominal intenso en flanco izdo, heces pastosas y aumento de los ruidos abdominales, que es valorado en atención primaria interpretando inicialmente el cuadro como posible gastroenteritis aguda. La exploración evidenciaba leve dolor a la palpación en hemiabdomen izdo y discreto aumento del peristaltismo. Ante la persistencia del dolor, se solicitan diversas pruebas complementarias.
Mientras está pendiente de nueva valoración, 10 días después de su visita a atención primaria, acude al servicio de Urgencias hospitalarias por presentar de nuevo episodio de intenso dolor abdominal en flanco izquierdo, acompañado en esta ocasión de diaforesis, nerviosismo y palidez cutánea. Valorado en tres ocasiones en servicios de urgencias, es diagnosticado y tratado como cólico renal. Presión arterial de 136/95 mm de Hg; frecuencia cardiaca: 89 l/min; temperatura: 37,7o; sudoroso. A la exploración abdominal presenta dolor en flanco izquierdo con sucusión renal izquierda positiva. El resto de la exploración física es normal. Las radiografías de tórax y abdomen son normales. Se objetiva mínima leucocitosis (10.500), leve hiponatremia y PCR=13.
Al no mostrar mejoría clínica con tratamiento sintomático se realiza ecografía abdomino-pélvica donde se evidencia una tumoración en glándula suprarrenal izquierda. Tras este resultado se pide TAC donde se objetiva una masa de 6,5 cm de diámetro dependiente de la glándula suprarrenal izquierda (Fig. 1) que plantea realizar diagnóstico diferencial entre feocromocitoma o carcinoma suprarrenal.
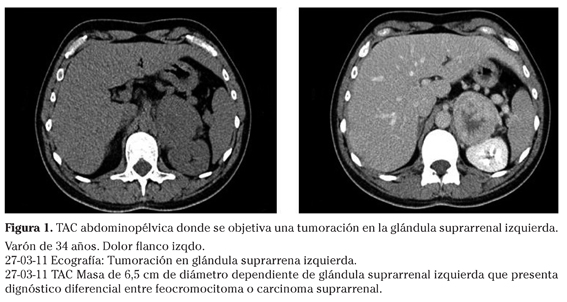
Tras este resultado se deriva al servicio de Urología y de éste al de Medicina Interna para estudio funcional, en el intento de descartar feocromocitoma. La determinación de metanefrinas y catecolaminas en orina muestra una lectura de más de cinco veces el valor normal para metanefrinas totales en orina y para catecolaminas libres urinarias. Se diagnostica de masa suprarrenal izquierda funcionante, con marcada elevación de metanefrinas por probable feocromocitoma. Una vez integrado el diagnóstico de feocromocitoma se decide practicar adrenalectomía total izquierda laparoscópica previo bloqueo con dosazoxina 4 mg/día. El análisis anatomopatológico de los hallazgos quirúrgicos demuestra que se trata de un feocromocitoma maligno de 130 g y 6 x 6 cm, con extensa necrosis, marcado pleomorfismo celular, abundantes mitosis atípicas (<50 x 10 cga), invasión capsular y un trombo tumoral en una vena de grueso calibre (la vena clampada), sin extensión extraadrenal. El periodo transoperatorio transcurre sin complicaciones. Durante todo el proceso el paciente presenta cifras tensionales normales con buen estado general, y no se lleva a cabo ningún tratamiento.
Discusión
En este caso el diagnóstico de feocromocitoma no se sospechó por haber hipertensión de difícil control en un paciente joven, que es la clínica más frecuente de presentación1-3. El cuadro debutó con dolor abdominal acompañado de diaforesis y nerviosismo, por el que se realizó una ecografía abdominal que evidenció una tumoración en glándula suprarrenal izquierda. Tras este resultado se realizó un TAC abdominal que describió la tumoración como una masa de 6,5 cm de diámetro dependiente de la glándula suprarrenal izquierda y que presentaba diagnóstico diferencial entre feocromocitoma y carcinoma suprarrenal.
La determinación de metanefrinas en orina de 24 horas con un aumento de más de cinco veces el valor normal, la prueba bioquímica más sensible por ser la menos afectada por medicamentos, confirmó el diagnóstico de feocromitoma. El diagnóstico de localización suele realizarse una vez confirmado el diagnóstico bioquímico, pero en este caso al cursar con una presentación menos frecuente, primero fueron las pruebas bioquímicas y posteriormente las de imagen. Al ser un tumor menor de 8 cm se eligió la resección por vía laparoscópica, ya que ofrece varias ventajas respecto a la cirugía convencional.
El pronóstico es bueno, excepto en los casos de enfermedad maligna, como resultó ser el de nuestro paciente, en los que la tasa de supervivencia a los cinco años es menor del 50%. La cirugía del feocromocitoma no siempre conduce a la curación de los pacientes, incluso con tumores benignos. Se han descrito recidivas en un 16% de los pacientes en algunas series. Así pues el seguimiento clínico y bioquímico de los pacientes debe ser indefinido.
Los tratamientos radiofarmacéuticos y citotóxicos de que se dispone para el tratamiento del feocromocitoma maligno, en el mejor de los casos son paliativos y rara vez curativos18. En bastantes casos los efectos secundarios no compensan los beneficios obtenidos.
Tal y como se ha señalado en otros estudios19,20, la predicción de la malignidad potencial de los fecocromocitomas es una tarea complicada y se encuentra todavía sin una respuesta definitiva.
Bibliografía
1. Petrina ME, Calderón DM, Menéndez EL. Feocromocitoma y paraganglioma. An Sist Sanit Navar 1998; 21: 31-46. [ Links ]
2. Bravo EL. Evolving concepts in the pathophysiology, diagnosis and treatment of Pheochromocytoma. Endocr Rev 1994; 15: 356-368. [ Links ]
3. Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful Pheochromocytoma operation. Hypertension 1997; 29: 1133-1139. [ Links ]
4. Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant A, Clive S et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab 2001; 86: 5210-5216. [ Links ]
5. Oleaga A, Goñi F. Feocromocitoma: actualización diagnóstica y terapéutica. Endocrinol Nutr 2008; 55: 202-216. [ Links ]
6. Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Ali A et al. A survey on adrenal incidentaloma in Italy. Study group in adrenal tumours of the Italian Society of Endocrinology. J Clin Endocrinol Metab 2000; 85: 637-644. [ Links ]
7. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346: 1459-1466. [ Links ]
8. Motta-Ramírez GA, Remer EM, Herts BR Gill IS, Hamrahian AH. Comparison of CT findings in symptomatic and incidentally discovered pheochromocytomas. AJR Am J Roentgenol 2005; 185: 684-688. [ Links ]
9. Lenders JW, Pacak K, Walter MM, Linehan WM, Mannelli M, Friberg P et al. Biochemical diagnosis of pheochromocytoma. Which test is best? JAMA 2002; 287: 1427-1434. [ Links ]
10. Bravo EL, Tagle R. Pheochromocytomas: State of the art and future prospects. Endocr Rev 2003; 24: 539-553. [ Links ]
11. Walther MM, Keiser HR, Linehan WM. Pheochromocytoma: evaluation, diagnosis, and treatment. World J Urol 1999; 17: 35-39. [ Links ]
12. Bravo EL. Evolving concepts in the pathophysiology, diagnosis and treatment of pheochromocytoma. Endocr Rev 1994; 15: 356-368. [ Links ]
13. Kebebew E, Siperstein AE, Clark OH, Duh QY. Results of laparoscopic adrenalectomy for suspected and unsuspectedmalignant adrenal neoplasms. Arch Surg 2002; 137: 948-951. [ Links ]
14. BjØrn E, Airazat MK, Tom M, Per F, Tonnessen TI, Fosse E. Laparoscopic and open surgery for pheochromocytoma. BMC Surgery 2001; 1: 2. [ Links ]
15. Tsuru N, Ushiyama T, Suzuki K. Laparoscopic adrenalectomy for primary and secondary adrenal tumours. J Endourology 2005; 19: 702-708. [ Links ]
16. Liao CH, Chueh SC, Lai MK, Hsiao PJ, Chen J. La paroscopic adrenalectomy for potentially malignant adrenal tumours greater than 5 cm. J Clin Endocrinol Metab 2006; 91: 3080-3083. [ Links ]
17. Tanaka S, Ito T, Tomoda J, Higashi T, Yamada G, Tsuji T. Malignant pheochromocytoma with hepatic metastasis diagnosed 20 years after resection of the primary adrenal lesion. Intern Med 1993; 32: 789-794. [ Links ]
18. Jiménez C, Cabanillas M, Santarpia L, Jonasch E, Kyle K, Lano E et al. Use of the tyrosine kinase inhibitor suitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pPheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab 2009; 94: 386-391. [ Links ]
19. Harari A, Inabnet III W. Malignant pheocromocytoma: a review. Am J Surg 2011; 201: 700-708. [ Links ]
20. Scholz T, Schulz C, Klose S, Lehnert H. Diagnostic management of benign and malignant pheochromocytoma. Exp Clin Endocrinol Diabetes 2007; 115: 155-159. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ana Arillo Crespo
Centro de Salud Chantrea
C/ San Cristóbal s/n
31015 Pamplona
E-mail: aarilloc@cfnavarra.es
Recepción: 14 de julio de 2011
Aceptación provisional: 21 de julio de 2011
Aceptación definitiva: 13 de octubre de 2011

