










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.36 no.3 Pamplona sep./dic. 2013
https://dx.doi.org/10.4321/S1137-66272013000300022
NOTAS CLÍNICAS
Acromegalia ectópica por carcinoide bronquial
Ectopic acromegaly due to a bronchial carcinoid
J. Rojo Álvaro, J.J. Pineda Arribas, E. Anda Apiñániz, L. Pérez García, J. Lafita Tejedor, Ll. Forga Llenas
Servicio de Endocrinología. Complejo Hospitalario de Navarra. Pamplona. Navarra.
Dirección para correspondencia
RESUMEN
Los tumores que causan acromegalia ectópica pueden hacerlo a través de la secreción de GH o de GHRH. Se han descrito un centenar de casos de acromegalia ectópica por secreción de GHRH. Dado la rareza de esta patología presentamos un caso clínico con el objetivo de aportar nuestra experiencia diagnóstico-terapéutica y de seguimiento posterior.
Se presenta el caso de una paciente con rasgos físicos acromegaloides de varios años de evolución. De forma concomitante también presentaba otros síntomas acompañantes sugestivos de posible origen bronquial. Ante la sospecha clínica de acromegalia se optó por confirmarlo bioquímicamente y posteriormente mediante estudio de imagen. Se descartó el origen hipofisario por lo que se realizó el despistaje de un tumor neuroendocrino bronquial y/o gastrointestinal por ser las localizaciones más frecuentes. El tratamiento de elección fue la resección quirúrgica.
Palabras clave: Acromegalia ectópica. GHRH. Carcinoide bronquial. Tumor neuroendocrino.
ABSTRACT
Tumours that cause ectopic acromegaly can do so through the secretion of GH or GHRH. A hundred cases of ectopic acromegaly due to secretion of GHRH have been described. Given the rarity of this pathology, we present a clinical case with the aim of contributing our diagnostic-therapeutic experience and the subsequent follow-up.
We present the case of a patient with acromegaloid physical features that had evolved over several years. Concomitantly, he also presented other accompanying symptoms that suggestive of a possible bronchial origin. Facing the clinical suspicion of acromegaly, we opted to confirm it biochemically and subsequently through image study. A hypophysary origin was ruled out, so we carried out screening for a bronchial neuroendocrine and/or gastrointestinal tumor as they are the most frequent localizations. The treatment of choice was surgical resection.
Key words: Ectopic acromegaly. GHRH ectopy. Bronchial carcinoid. Neuroendocrine tumor.
Introducción
La secreción hormonal ectópica se conoce desde hace más de 80 años. En 1941, Fuller Albright, considerado el padre de la endocrinología moderna, propuso la idea de que los tumores pueden causar síndromes endocrinos secretando hormonas de forma inapropiada. GW Liddle, en 1969 acuñó el término «síndrome hormonal ectópico» para referirse a este tipo de situaciones1.
La acromegalia generalmente se debe a la hipersecreción de hormona de crecimiento (GH) por un adenoma hipofisario benigno. Se estima una incidencia de 3 casos/millón de habitantes/año y una prevalencia de 60 casos/millón. Los tumores que causan acromegalia ectópica pueden hacerlo a través de la secreción de GH o de hormona liberadora de hormona de crecimiento (GHRH). Desde el descubrimiento de la GHRH en 1982, tan solo se han descrito un centenar de casos de acromegalia ectópica por secreción de GHRH2.
A continuación presentamos un caso de acromegalia ectópica por GHRH con el objetivo de aportar nuestra experiencia diagnóstico-terapéutica.
Caso clínico
Mujer de 38 años de edad que acudió remitida a consulta por sospecha de acromegalia desde el servicio de Reumatología al que había acudido por dolores articulares generalizados. La paciente refería que desde hace 8 años había notado aumento del tamaño de la nariz y ambos arcos cigomáticos. También refería engrosamiento de los dedos de las manos y aumento de hasta dos números de zapato. Así mismo refería separación progresiva de los dientes de la arcada inferior, astenia, aumento de sudoración sobre todo en las manos, la piel más engrosada, seca y tosca, la voz más grave y ser roncadora habitual.
Había tenido dos embarazos 5 y 2 años antes de la consulta sin ninguna dificultad para la concepción ni para la lactancia. Desde el segundo embarazo continuaba con galactorrea a la expresión. Posteriormente las menstruaciones eran normales, pero había estado un año en amenorrea tras el segundo parto.
Interrogada no refería cefaleas ni alteraciones visuales. Comentaba presentar sensación de inestabilidad y mareo con el ortostatismo de años de evolución sin cambios. Desde hace dos años aproximadamente presentaba cuadros autolimitados con una frecuencia quincenal, de sensación de calor e hipersudoración facial y torácica que cedían en minutos. Refería taquicardia y sensación de palpitaciones con la actividad física pero no en reposo. No había presentado disnea. Habitualmente tenía estreñimiento pertinaz sin restos patológicos.
No tenía antecedentes personales ni familiares de interés, así como tampoco hábitos tóxicos.
En la exploración física destacaban signos físicos acromegaloides con ambos arcos cigomáticos prominentes, diastema, la piel gruesa elefantiásica, las manos toscas, ensanchadas, con engrosamiento de las articulaciones y los pies también ensanchados.
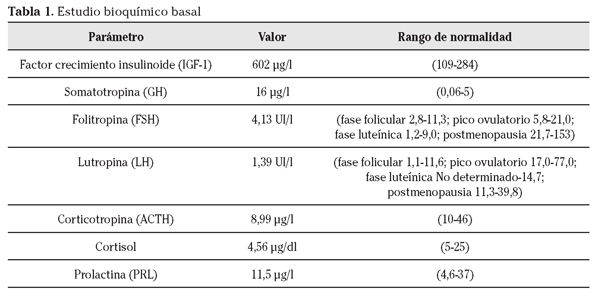
Con la sospecha clínica de acromegalia se solicitó estudio bioquímico basal con los resultados mostrados en la Tabla 1. Tras la obtención de los mismos se decidió realizar la determinación de la secreción de GH tras sobrecarga oral de glucosa (SOG) con 75 gramos (g), obteniendo los resultados mostrados en la Tabla 2.

Una vez confirmada la sospecha bioquímica de acromegalia se solicitó una resonancia nuclear magnética (RMN) craneal que mostraba el área hipotálamo-hipofisaria sin alteraciones.
Ante la falta de disponibilidad de determinar la GHRH en España y con la sospecha de que pudiera tratarse de una acromegalia ectópica se solicitó una radiografía (Rx) de tórax donde se apreciaba una imagen nodular hiperdensa en hilio derecho (Fig. 1A).

Posteriormente se realizó una tomografía axial computarizada (TAC) torácica que confirmaba la presencia de una imagen hiliar derecha ovalada de 7 x 4 x 3,5 cm con densidad sólida adyacente al mediastino (Fig. 1B).
Para completar el estudio se realizó un octreoscan que mostró una hipercaptación del hilio pulmonar derecho (Fig. 2).

Así mismo se realizaron otros estudios complementarios: cromogranina A >1200 ng/ml (19,4-98,1), gastrina 290 ng/l (13-115), glucagón 82 pg/ml (59-177), péptido intestinal vasoactivo 11,5 pmol/l (0-30), polipéptido pancreático 17,1 pmol/l (0-0), enolasa neuronal específica 6,7 µg/l (0-12,5), serotonina 491 ng/ml (80-450), 5-hidroxiindolacetato en orina 11,6 mg/24 horas (2-10), ecocardiograma y colonoscopia normales, TAC cérvico-tóraco-abdominal sin otras alteraciones.
Con el diagnóstico de sospecha de acromegalia ectópica por tumor carcinoide pulmonar probablemente secretor de GHRH fue intervenida practicando una lobectomía derecha. El estudio anatomo-patológico confirmó que se trataba de un carcinoma neuroendocrino de 5 x 3,5 cm moderadamente diferenciado pT2NxM0. En el estudio inmunohistoquímico las células tumorales resultaron positivas para sinaptofisina, cromogranina y el marcador CD 56. No disponemos de determinación de GHRH.
Se completó tratamiento adyuvante con radioterapia externa (dosis 60 Gray).
En revisiones posteriores los marcadores bioquímicos se normalizaron: cromogranina A 80 ng/ml, IGF-1 136 µg/l, GH 0,36 µg/l y el TAC torácico de control fue normal, mostrando únicamente secuelas postquirúrgicas.
Tras 5 años de seguimiento los datos bioquímicos y radiológicos no han mostrado evidencia de enfermedad y la paciente permanece clínicamente asintomática.
Discusión
Los tumores que causan acromegalia ectópica pueden hacerlo a través de la secreción de GH o de GHRH.
Los criterios clínicos para establecer el diagnóstico de secreción hormonal ectópica son los que se exponen a continuación. Deben cumplirse el primero y por lo menos otro de los siguientes3:
1) Un síndrome clínico y bioquímico de exceso hormonal se asocia con una neoplasia.
2) Los niveles de hormona en sangre y orina están inapropiadamente elevados junto con una producción hormonal endógena normal o suprimida.
3) Se han excluido otros posibles mecanismos causales.
4) Se demuestra la presencia de ácido ribonucleico (RNA) mensajero de la hormona y el correspondiente producto hormonal en las células tumorales y, por tanto, la producción hormonal es mayor en el tejido tumoral que en los tejidos de alrededor.
5) El síndrome revierte al resecar el tumor y, en cambio, persiste al resecar la glándula endocrina habitualmente responsable del síndrome de hipersecreción hormonal.
6) Existe un gradiente arteriovenoso de la concentración hormonal a través del tumor. En cuanto a la etiopatogenia de los síndromes hormonales paraneoplásicos, es probable que representen la confluencia de diversos mecanismos genéticos y epigenéticos. Entre ellos, se ha propuesto que las mutaciones que subyacen en las células tumorales pueden ser responsables no sólo del inicio de la transformación neoplásica sino también de la activación o reexpresión de genes responsables de la producción hormonal4.
Adicionalmente, otros acontecimientos que afectan a los genes tales como la metilación, pueden ser la causa del desarrollo de estos síndromes5.
Se conocen dos entidades bien documentadas de acromegalia originada por hipersecreción de GH de origen no hipofisario, el tumor de células de islotes pancreáticos y el linfoma no-Hodgkin6. La acromegalia debida a secreción ectópica de GHRH, aunque más frecuente, es también rara, con una prevalencia < 0,5 % de todos los casos de acromegalia. Los carcinoides bronquiales y gastrointestinales, seguidos por las neoplasias de células de los islotes pancreáticos son los que con más frecuencia causan excesiva secreción de GHRH7. Son más frecuentes en el sexo femenino, con una edad media de 40 años e historia de unos 10 años de evolución de la clínica acromegálica y afectando predominantemente el pulmón derecho con un tamaño medio de 1-2 cm8.
La presencia de clínica adicional como disnea, pitidos torácicos, flushing facial, dispepsia y cálculos renales puede ayudar para sospechar patología extrahipofisaria. Si no está disponible la determinación de GHRH ante la ausencia de imagen compatible con adenoma hipofisario puede realizarse un TAC torácico y una ecografía abdominal para excluir la posibilidad de un síndrome ectópico de GHRH9.
Se han descrito otras etiologías menos habituales como tumores carcinoides tímicos asociados a una neoplasia endocrina múltiple tipo 1 (MEN-1)10, casos de hiperplasia difusa idiopática de células neuroendocrinas pulmonares con hipersecreción de GHRH y ACTH11, adenomas extrahipofisarios ubicados en los senos carvernosos12 y un caso asociado a un paraganglioma mediastínico2. En los casos debidos a tumores pancreáticos debe estudiarse de forma sistemática mutaciones de MEN-113.
Los tumores que secretan GHRH ocasionan secundariamente hipersecreción de GH hipofisaria. La anatomía patológica muestra habitualmente hiperplasia hipofisaria14. Analíticamente se detectan valores séricos elevados de GH, IGF-1 y también de GHRH. El 50% de los casos pueden presentar metástasis a pesar de lo cual el pronóstico generalmente es bueno. Se recomienda el tratamiento mediante abordaje quirúrgico del tumor primario causante del síndrome. Cuando la resección completa es posible, en la mayoría, se consigue remisión a largo plazo. En estos casos la concentración de GHRH se normaliza y su incremento es un certero indicador de recurrencia13. No obstante, los análogos de somatostatina representan una opción terapéutica útil en los pacientes no curados puesto que son capaces de mejorar la producción hormonal ectópica (54-70%) y la secreción hipofisaria de GH además de disminuir el tamaño tumoral10.
Bibliografía
1. Forga L, Anda E, Martínez JP. Síndromes hormonales paraneoplásicos. An Sist Sanit Navar 2005; 28: 213-226. [ Links ]
2. Ghazi A, Amirbaigloo A, Dezfooli AA, Saadat N, Ghazi S, Pourafkari M et al. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine 2013; 43: 293-302. [ Links ]
3. Bollanti L, Riondino G, Strollo F. Endocrine paraneoplastic syndromes with special reference to the elderly. Endocrine 2001; 14: 151-157. [ Links ]
4. DeLellis RA, Xia L. Paraneoplastic endocrine syndromes: a review. Endocrine Pathol 2003; 14: 303-317. [ Links ]
5. Newell-Price J. Propiomelanocortin gene expression and DNA methylation: implications for Cushing´s síndrome and beyond. J Endocrinol 2003; 177: 365-372. [ Links ]
6. Chanson P, Salenave S. Acromegaly review. Orphanet J Rare Dis 2008; 3: 1-17. [ Links ]
7. Garby L, Caron P, Claustrat F, Chanson P, Tabarin A, Rohmer V et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of Growth Hormone-Releasing Hormone (GHRH): A French nationwide series of 21 cases. J Clin Endocrinol Metab 2012; 97: 2093-2104. [ Links ]
8. Bolanowski M, Schopohl J, Marciniak M, Rzeszutko M, Zatonska K, Daroszewski J et al. Acromegaly due to GHRH-secreting large bronchial carcinoid. Complete recovery following tumor surgery. Exp Clin Endocrinol Diabetes 2002; 110: 188-192. [ Links ]
9. Gola M, Doga M, Bonadonna S, Mazziotti G, Vescovi PP, Giustina A. Neuroendocrine tumors secreting growth hormone-releasing hormone: Pathophysiological and clinical aspects. Pituitary 2006; 9: 221-229. [ Links ]
10. Boix E, Picó A, Pinedo R, Aranda I, Kovacs K. Ectopic growth hormone-releasing hormone secretion by thymic carcinoid tumour. Clin Endocrinol (Oxf) 2002; 57: 131-134. [ Links ]
11. Fessler MB, Cool CD, Miller YE, Schwarz MI, Brown KK. Idiopathic diffuse hyperplasia of pulmonary neuroendocrine cells in a patient with acromegaly. Respirology 2004; 9: 274-277. [ Links ]
12. Mitsuya K, Nakasu Y, Nioka H, Nakasu S. Ectopic growth hormone releasing adenoma in the cavernous sinus. Neurol Med Chir (Tokyo) 2004; 44: 380-385. [ Links ]
13. Borson-Chazot F, Garby L, Raverot G, Claustrat F, Raverot V, Sassolas G. Acromegaly induced by ectopic secretion of GHRH: A review 30 years after GHRH discovery. Ann Endocrinol (Paris) 2012; 73: 497-502. [ Links ]
14. Shoback D, Funk J. Humoral manifestations of malignancy. En: Greenspan FS y Gardner DG Editores. Basic and Clinical Endocrinology. New York Lange Medical Books/Mc Graw-Hill 1997; 778-791. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Jorge Rojo Álvaro
Servicio de Endocrinología
Complejo Hospitalario de Navarra
C/ Irunlarrea, no 3
31008, Pamplona
jorgerojo23@hotmail.com
Recepción: 26 de febrero de 2013
Aceptación provisional: 22 de abril de 2013
Aceptación definitiva: 3 de mayo de 2013

