










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
Print version ISSN 1137-6627
Anales Sis San Navarra vol.37 n.1 Pamplona Jan./Apr. 2014
https://dx.doi.org/10.4321/S1137-66272014000100020
Pseudoobstrucción intestinal crónica por displasia neuronal intestinal tipo B (DNI B). A propósito de un caso
Chronic intestinal pseudo-obstruction due to intestinal neuronal dysplasia type B (IND B), concerning one case
S. Junquera Bañares1, E. Oria Mundín1, A. Córdoba Iturriagagoitia2, J.I. Botella-Carretero3
1. Servicio de Endocrinología y Nutrición. Complejo Hospitalario de Navarra
2. Servicio de Anatomía patológica. Complejo Hospitalario de Navarra
3. Servicio de Endocrinología y Nutrición. Hospital Ramón y Cajal. Madrid
Dirección para correspondencia
RESUMEN
La displasia neuronal intestinal tipo B (DNIB) es una enfermedad poco frecuente debida a la hiperplasia del plexo submucoso parasimpático que cursa con alteración de la motilidad intestinal, ocasionando síntomas de estreñimiento y cuadros suboclusivos1. Es una enfermedad difícil de diagnosticar que requiere alta sospecha clínica y debería incluirse en el diagnóstico diferencial de los pacientes con cuadros suboclusivos de repetición con el fin de hacer un diagnóstico correcto temprano y evitar complicaciones derivadas de cirugías innecesarias que empeoran el pronóstico.
Se presenta el caso de un varón marroquí de 33 años que fue ingresado en nuestro hospital en 2 ocasiones un total de 11 meses, precisando durante 5 meses nutrición parenteral total (NPT). El análisis inmunohistoquímico de la boca de ileostomía y colostomía lleva al diagnóstico de DNIB. Dieciocho meses después, el paciente hace vida normal y ha recuperado los 25 kilos perdidos siguiendo las indicaciones dietéticas y con los suplementos enzimáticos.
Palabras clave. Displasia neuronal intestinal B. Suboclusión intestinal.
ABSTRACT
Intestinal neuronal dysplasia type B (IND B) is an infrequent disease due to hyperplasia of the parasympathetic submucous plexus which causes alteration of intestinal motility, giving rise to symptoms of constipation and subocclusive manifestations. The disease is difficult to diagnose. It requires high clinical suspicion and should include differential diagnosis of patients with repeated subocclusive manifestations in order to make an early and correct diagnosis and avoid complications derived from unnecessary surgery that worsens the prognosis.
We present the case of a 33-year-old Moroccan male who was admitted to our hospital on 2 occasions in 11 months, requiring total parenteral nutrition (TPN) for five months. The immunohistochemical analysis of the ileostomy and colostomy stoma led to a diagnosis of IND B. Eighteen months later, the patients is leading a normal life and has recovered the 25 kilos lost following the dietary indications and with the enzymatic supplements.
Key words. Intestinal neuronal dysplasia B. Intestinal subocculsion.
Introducción
El síndrome de pseudoobstrucción intestinal crónico (POIC) se caracteriza por la presencia de cuadros clínicos recidivantes que simulan una obstrucción mecánica del intestino delgado o grueso en ausencia de lesiones anatómicas que obstruyen el flujo del contenido intestinal. Se origina como consecuencia de una alteración de la motilidad intestinal que no puede hacer progresar su contenido debido a la afectación de su componente muscular, nervioso o de ambos. Esto puede ser debido a un proceso primario o secundario a una enfermedad sistémica1.
La displasia neuronal intestinal (DNI) es una neuropatía primaria poco frecuente que causa POIC y fue descrita por primera vez en 1971 en niños por Meier-Ruge2. En 1983 Fadda y Meier-Ruge diferencian la tipo A y tipo B. La DNI A aparece en el 5% de los casos, es una aplasia congénita o hipoplasia de la inervación simpática que se presenta como una colitis grave y es exclusiva de la población pediátrica. La DNI B corresponde al 95% de los casos; es una hiperplasia del plexo submucoso parasimpático que ocasiona síntomas de estreñimiento de intensidad variable en niños y adultos3,4. Histológicamente la enfermedad se caracteriza por hiperganglionosis, presencia de ganglios gigantes intramurales, ganglios ectópicos y aumento de acetilcolinesterasa en lámina propia4. Es una entidad poco frecuente, subdiagnosticada y determina una alta morbimortalidad. Su diagnóstico es habitualmente difícil, suele demorarse muchos años después del inicio de los síntomas, los pacientes son etiquetados de funcionales en muchas ocasiones, y se les practica cirugías innecesarias que empeoran su pronóstico.
Caso clínico
Varón de 33 años, natural de Marruecos, y que reside en España de forma intermitente por motivos laborales. Fue trasladado desde su país al servicio de Urgencias de nuestro hospital por dolor abdominal y vómitos. El paciente refería haber estado ingresado los dos meses previos debido a un cuadro suboclusivo que no respondió a tratamiento médico. En el informe que aportó constaba que se le practicó enterolisis después de un mes de ayuno terapéutico sin observarse mejoría. En el postoperatorio presentó retraso del vaciado gástrico e íleo prolongado, por lo que tuvo que ser reintervenido. Se le hizo una colectomía derecha ampliada, anastomosis gastroyeyunal, ileostomía lateral derecha y colostomía lateral izquierda. En el informe anatomopatológico constaba que en los 14 cm de íleon y 140 cm de colon que se le extirparon, se observaron unas zonas de intestino retráctiles y otras muy dilatadas, con zonas de mucosa inflamadas y otras con extensa necrosis, además de la existencia de fibrosis peritonítica extensa y múltiples adherencias, pero no encontraron lesiones específicas. Después de dos meses de ayuno total con fluidoterapia iv por intolerancia a la nutrición enteral (NE), y habiendo perdido más de 25 kilos de peso, el paciente solicitó el alta voluntaria para trasladarse a España.
El paciente refería antecedentes de cuadros suboclusivos desde la infancia resueltos con tratamiento médico y varias intervenciones quirúrgicas: laparoscopia con resección de vólvulo a los 13 años, resección duodenal con gastroenteroanastomosis a los 29 años, y drenaje de absceso perihepático con afectación de pared intercostal por probable perforación duodenal de etiología no filiada a los 30 años. Entre los antecedentes familiares destacaba un hermano fallecido a los 39 años en una cirugía abdominal urgente (supuestamente por cáncer de colon sin ser documentado por autopsia) e hijo de 8 años con cuadros suboclusivos similares a los que presenta el paciente desde su infancia.
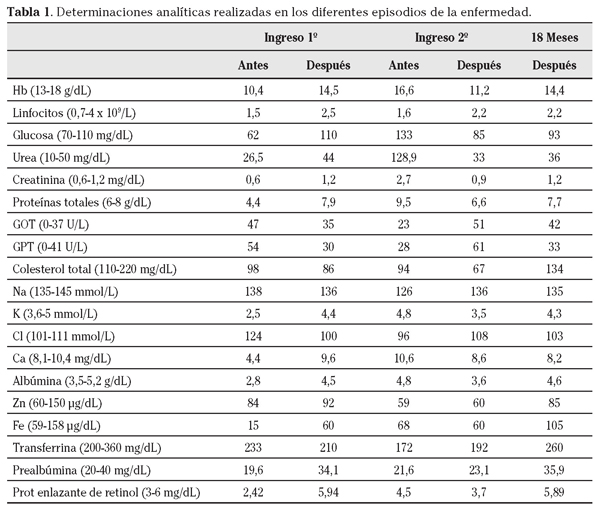
En la exploración física presentaba mal estado general, desnutrición severa y anasarca, y edemas con fóvea hasta rodillas. Abdomen blando y depresible con molestias difusas a la palpación y peristaltismo ausente. No signos de irritación peritoneal. Dos estomas sin contenido en línea de laparatomía media. Peso 58,6 Kg (IMC 16,2 Kg/m2). El resto de la exploración y pruebas complementarias fue normal, salvo los parámetros analíticos (Tabla 1).
Fue ingresado en el servicio de Cirugía; se le colocó una sonda de aspiración, se pautó antieméticos y procinéticos, y se comenzó a renutrir progresivamente con nutrición parenteral total (NPT). Se mantuvo la NPT durante dos meses hasta que se pasó a NE continua, y finalmente, se pudo retirar la sonda de aspiración e introducir nutrición oral con ayuda de procinéticos y medidas higiénico-dietéticas favorecedoras del peristaltismo. A las 14 semanas del ingreso se le dio de alta con un peso de 79 Kg (IMC 21,9 Kg/m2).
Tres meses después, el paciente vuelve a ingresar en el servicio de Cirugía por un cuadro de deshidratación hiponatrémica severa debido al aumento del débito por ileostomía de unos 3-4 litros/día e intolerancia oral de varios días de evolución; episodio que fue relacionado con un cuadro suboclusivo según los resultados de las pruebas complementarias que se le hicieron (Fig. 1). Se le corrigieron las alteraciones hidroelectolíticas que presentaba, y ante la intolerancia oral, se comenzó al tercer día del ingreso la NPT y se mantuvo los tres meses siguientes.

En el cuarto mes del segundo ingreso, al paciente se le realizó una enterolisis de todo el paquete intestinal, se desmontó la gastroenteroanastomosis previa, se resecó el intestino delgado a unos 40-50 cm del ángulo de Treiz y se extirparon 80 y 130 cm de íleon de sendos extremos, se liberó el colon y se realizó una anastomosis íleo-cólica latero-lateral mecánica. Dos meses después, tuvo que ser reintervenido por mal paso en anastomosis, se resecó la zona anastomótica anterior con la extirpación de 12 cm de íleon y se le realizó una nueva anastomosis íleo-cólica termino-lateral manual. En el análisis anatomopatológico de la boca de ileostomía y colostomía se observó hiperganglionosis en plexos parasimpáticos, aumento de ganglios inmaduros bcl2+ y de linfocitos intraepiteliales, junto con disminución de células intersticiales de Cajal, todo ello compatible con DNIB (Fig. 2). A los 8 meses se le dio de alta con síndrome de malabsorción y esteatorrea crónica secundaria a intervenciones previas, se pautaron medidas dietéticas y enzimas pancreáticos en cada ingesta, junto con un antidiarreico y resíncolestiramina a demanda. A los 18 meses acude a revisión con excelente estado general, hace vida normal, sigue los consejos dietéticos que se le indicaron, hace 2-3 deposiciones diarias no esteatorreicas, toma 4-6 comprimidos al día de suplementos enzimáticos (Kreon®10000) y su peso es 90,5 kilos (IMC 25,1 Kg/m2).

Discusión
Los antecedentes familiares del paciente y la afectación difusa de todo el intestino, son datos que orientan hacia una probable causa genética; pero hasta ahora no se sabe el tipo de herencia ni las mutaciones implicadas en la patogénesis de la DNI B.
Se cree que el protooncogen RET pudiera estar involucrado en la patogénesis de la enfermedad, pero por mecanismos moleculares diferentes a la enfermedad de Hirschsprung (EH). Se ha detectado una distribución característica de determinados polimorfismos en los pacientes afectados. Los rasgos clínicos de la DNI B recuerdan en algún aspecto a la EH, y aunque no hay regiones de agangliosis como en ésta, sí se ven en ambas enfermedades un aumento de fibras nerviosas extrínsecas en el intestino afectado. Además, se ha observado que hasta en un 25-35% de los pacientes con EH tienen regiones próximas al segmento agangliónido con características típicas de DNI B5.
Algunas hipótesis refieren mutaciones del gen EDNR36z y una disminución de Pten en ganglios de plexos entéricos4. Por otra parte, ciertas observaciones apuntan que la DNI B podría también tener un origen adquirido y ser causada por la reacción del sistema nervioso entérico a la obstrucción intestinal o enfermedades inflamatorias que podrían darse en el periodo fetal o postnatal6. Además, se puede ver ocasionalmente hiperplasia de las células ganglionares similar a DNI B en pacientes que sufren estreñimiento crónico, en enfermedades inflamatorias como colitis ulcerosa, isquémica o enfermedad de Crohn, o en zonas contiguas a segmentos agangliónicos en EH. Las anomalías que se ven en el plexo submucoso y entérico pueden ser debidas a que las células nerviosas funcionantes pueden hiperactivarse (hiperplasia e hipertrofia) para estimular al plexo anormal, resultando la hiperplasia de células ganglionares7. Sin embargo, en las formas adquiridas, no suele haber una afectación difusa de todo el intestino como es frecuente observar en las formas de causa genética.
El diagnóstico de esta enfermedad es difícil y se puede demorar una media de 8 años después de comenzar los síntomas y hasta un 88% de los pacientes han sido intervenidos previamente una media de 2,96 veces de forma innecesaria, según se observa en la serie de mayor número de pacientes con POIC8. El caso que se expone es un ejemplo de ello, ya que comenzó con síntomas desde su infancia, y antes de acudir a nuestro hospital se le había intervenido al menos en cuatro ocasiones.
Los signos radiológicos no diferencian esta enfermedad de otro cuadro obstructivo de diferente etiología, apreciándose asas dilatadas y niveles hidroaéreos en bipedestación. Un 20-30% de las pruebas radiológicas pueden ser normales o con alteraciones poco evidentes, particularmente en las obstrucciones altas de yeyuno proximal3. A veces se dan alteraciones segmentarias, como en los casos de megaduodeno, pero generalmente suelen afectar al intestino delgado y/o colon de forma global.
Para llegar al diagnóstico es preciso el análisis de todo el espesor de la pared mediante técnicas inmunohistoquímicas utilizando los marcadores siguientes: bcl-2 para células ganglionares inmaduras, c-kit para células intersticiales de Cajal, GFAP proteína glial fibrilar ácida para plexos nerviosos alrededor de vasos sanguíneos y S-100 y sinaftofina para ver el tamaño de los ganglios entéricos y la inervación neuromuscular. Por el contrario, en el análisis histopatológico realizado de rutina con tinción de hematoxilina-eosina no se detectan anomalías estructurales en mucosa, submucosa y capa neuromuscular del intestino, motivo por el cual esta enfermedad pasa desapercibida en muchas ocasiones.
Los criterios diagnósticos según Meier-Ruge son la existencia de ganglios gigantes en la submucosa (2 a 3 veces el tamaño normal), con más de 7 células ganglionares por ganglión, hiperplasia del plexo submucoso, aumento de la actividad acetilcolinesterasa en las fibras nerviosas de la lámina propia y adventicia de las arterias submucosas, y células nerviosas heterotópicas en la muscular de la mucosa y en la lámina propia. Hay autores que consideran suficiente para el diagnóstico la hiperplasia del plexo submucoso y el aumento de la actividad acetilcolinesterasa alrededor de los vasos sanguíneos; y para otros, es suficiente la existencia de hiperganglionosis y ganglios gigantes. Estos hallazgos pueden acompañarse o no de un infiltrado inflamatorio difuso en intestino delgado. La DNI B es una anomalía primaria del plexo submucoso, pero también se pueden detectar cambios hiperplásicos en el plexo mientérico como se observa en nuestro paciente4.
El tratamiento de la forma aguda se basa en la colocación de una sonda nasogástrica cuando el paciente no tolera ni sus propias secreciones y suministro de procinéticos. En los pacientes con gastroparesia se utiliza eritromicina iv antes de las comidas en la forma aguda y crónica. El tratamiento de soporte se basa en modificaciones dietéticas y medicamentos procinéticos, pero en casos refractarios necesitan NPT de forma crónica y, a veces, se plantea el trasplante intestinal si aparecen complicaciones hepáticas, infecciosas o trombóticas graves asociadas a la NPT.
La cirugía se utiliza para resecar la enfermedad localizada en intestino delgado o el colon en caso de estreñimiento o para tener un acceso al estómago o intestino delgado para descomprimir o mejorar síntomas. En dos series quirúrgicas de pacientes adultos intervenidos por cuadro de POIC se vio que los pacientes con enfermedad localizada tenían una evolución satisfactoria después de la cirugía, a diferencia de los que tenían enfermedad difusa (la mayoría por DNIB), que seguían con clínica en el seguimiento un tercio de los mismos9,10. Sin embargo, la resección de la enfermedad localizada debe hacerse con cautela porque la experiencia clínica demuestra con frecuencia que en muchos casos la enfermedad es difusa o la cirugía se asocia al desarrollo de adherencias que facilitan la recurrencia de la sintomatología.
La evolución y el pronóstico en los niños es más favorable que en adultos, normalizándose la motilidad intestinal con el tiempo hasta en los dos tercios de los casos, por lo que se cree que esta enfermedad puede observarse de forma transitoria y tratarse de una inmadurez del plexo mientérico o submucosa más que de una malformación del sistema nervioso autónomo per se11.
Sin embargo, su aparición en adultos es muy poco frecuente y el pronóstico suele ser malo. En 1989 Stoss y col ya describieron esta enfermedad en adultos con estreñimiento crónico y diverticulosis en el colon sigmoide4; y en 1994 publicaron una serie de 41 paciente con POIC, de los cuales, 24 tenían criterios histológicos de DNIB12. Posteriormente, son anecdóticos los casos publicados de DNIB, y solo hay uno similar al que exponemos, con afectación ileal y que tuvo un desenlace fatal por isquemia masiva del intestino delgado4. Afortunadamente nuestro paciente, después de 11 meses de ingreso hospitalario y 5 meses con NPT, dos años después hace una vida normal siguiendo una dieta especia y sin necesidad de ningún suplemento nutricional, ha recuperado el peso perdido.
La mortalidad por POIC ha pasado a ser del 30% en las series de hace 25 años a un 11% en las actuales gracias a los avances en la nutrición8. Es fundamental el manejo multidisciplinar (anatomopatólogos, gastroenterólogos, cirujanos y nutricionistas) de estos enfermos y tener una sospecha clínica para poder hacer un diagnóstico correcto lo más precozmente posible y evitar complicaciones innecesarias que empeoran el pronóstico de los mismos.
Agradecimientos
Agradecemos al Dr Alejandro Adrián Recalde (in memorian) el magnífico trabajo realizado, la dedicación y el gran empeño que puso para que este paciente saliera adelante.
Bibliografía
1. Muñoz MT, Solís Herruzo JA. Pseudo-obstrucción intestinal crónica. Rev Esp Enferm Dig 2007; 99:100-111. [ Links ]
2. Meier-Ruge WA, Bruder E, Kapur RP. Intestinal neuronal dysplasia type B: one giant ganglion is not good enough. Pediatr Dev Pathol 2006; 9: 444-452. [ Links ]
3. Soto Castillo D, Bannura Cumsille G, Peñaloza Montecinos P. Displasia neuronal intestinal como causa de constipación crónica pertinaz en adulto. Rev Chilena de Cirugía 2006; 58: 54-58. [ Links ]
4. Vijayaraghavan R, Chandrashekar R, Melkote Jyotiprakash A, Kumar R, Rashmi MV, Shanmukhappa Belagavi C. Intestinal neuronal dysplasia (type B) causing fatal small bowel ischaemia in an adult: a case report. Eur J Gastroenterol Hepatol 2006; 18: 773-776. [ Links ]
5. Fernández RM, Sánchez-Mejías A, Ruiz-Ferrer MM, López-Alonso M, Antiñolo G, Borrego S. Is the RET proto-oncogene involved in the pathogenesis of intestinal neuronal dysplasia type B? Mol Med Report 2009; 2: 265-270. [ Links ]
6. Sánchez-Mejías A, Fernández RM, Antiñolo G, Borrego S. A new experimental approach is required in the molecular analysis of intestinal neuronal dysplasia type B patients. Exp Ther Med 2010; 1: 999-1003. [ Links ]
7. Jeng YM, Mao TL, Hsu WM, Huang SF, Hsu HC. Congenital interstitial cell of cajal hyperplasia with neuronal intetsinal dysplasia. Am J Surg Pathol 2000; 24: 1568-1572. [ Links ]
8. Stanghellini V, Cogliandro RF, De Giorgio R et al. Natural history of chronic idiopathic intestinal pseudo-obstruction in adults: a single center study. Clin Gastroenterol Hepatol 2005; 3: 449-458. [ Links ]
9. Han EC, Oh HK, Ha HK, Choe Ek, Moon SH, Ryoo SB et al. Favorable surgical treatment outcomes for chronic constipation with features of colonic psedo-obstruction. World J Gastroenterol 2012; 18: 4441-4446. [ Links ]
10. Choe EK, Park SH, Park KJ. Colonic pseudo-obstruction with distinct transitional zone in adult constipation patients: pathological analysis and results of surgical treatment. Am Surg 2011; 77: 736-742. [ Links ]
11. Schimpl G, Uray E, Ratschek M, Höllwarth ME. Constipation and intestinal neuronal dysplasia type B: a clinical follow-up study. J Pediatr Gastroenterol Nutr 2004; 38: 308-311. [ Links ]
12. Stoss F, Meier-Ruge W. Experience with neuronal intestinal dysplasia (NID) in adults. Eur J Pediatr Surg 1994; 4: 298-302. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Sonia Junquera Bañares
Servicio de Endocrinología y Nutrición
Complejo Hospitalario de Navarra
C/ Irunlarrea, 3
31008 Pamplona
E-mail: sojuba@hotmail.com
Recepción: 1 de noviembre de 2013
Aceptación provisional: 25 de noviembre de 2013
Aceptación definitiva: 28 de noviembre de 2013

