










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.38 no.1 Pamplona ene./abr. 2015
https://dx.doi.org/10.4321/S1137-66272015000100009
Perfil neuropsicológico de la degeneración lobar frontotemporal
Neuropsychological profile of frontotemporal lobar degeneration
M.M. Carmona-Abellán, M.R. Luquin, I. Lamet, N. Pujol Giménez, P. Esteve-Belloch, M. Rivero
Clínica Universidad de Navarra. Departamento de Neurología. Pamplona
Ninguno de los autores tiene conflicto de intereses.
Dirección para correspondencia
RESUMEN
La degeneración lobar frontotemporal engloba tres síndromes diferentes, que comparten características clínicas y patológicas comunes, dificultando así su diagnóstico en estadios iniciales. Se incluyen en este grupo las tres variantes de la demencia frontotemporal, el síndrome corticobasal y el síndrome de parálisis supranuclear progresiva. Se ha llevado a cabo una revisión del perfil neuropsicológico de cada uno de los síndromes, que permita clarificar las características fundamentales que los definen y ayudar a diferenciarlos de otras demencias.
Se ha hecho una revisión de los diferentes trabajos publicados en la literatura al respecto, describiendo las características clínicas, patológicas y los hallazgos de imagen fundamentales de cada entidad para describir de manera exhaustiva los hallazgos en los diferentes dominios neuropsicológicos y su progresión.
Aunque existe un solapamiento entre los síndromes que conforman la degeneración lobar frontotemporal, la comparación del perfil neuropsicológico de las mismas entre sí y frente a otras demencias permite establecer características propias de su perfil neuropsicológico para llevar a cabo un diagnóstico diferencial.
Palabras clave: Demencia frontotemporal. Neuropsicología. Dominios cognitivos. Parkinsonismos. Test neuropsicológico.
ABSTRACT
Frontotemporal lobar degeneration encompasses three different syndromes, with clinical and pathologic commonalities, making diagnosis difficult in early stages. Three subtypes are recognized: frontotemporal dementia and its three variants, corticobasal syndrome and supranuclear palsy syndrome. The objective of this study is to review the neuropsychological features of each syndrome in order to differentiate amongst subtypes as well as from other forms of dementia.
We review multiple studies from the literature, highlighting the main clinical features, neuropathology and changes in brain imaging of each syndrome. Subsequently, we describe the neuropsychological profile compared to other dementias, and how it progresses over time.
Although there is an overlap amongst the different subtypes of frontotemporal lobar degeneration, neuropsychological profiles can help identify subtypes and discriminate frontotemporal lobar degeneration from other forms of dementia.
Key words: Frontotemporal dementia. Neuropsychology. Cognitive Domains. Parkinsonism. Neuropsychological test.
Introducción
La degeneración lobar frontotemporal (DLFT) es un término que engloba un grupo heterogéneo de enfermedades neuropatológicamente caracterizado por la presencia de inclusiones proteicas anormales en el citoplasma o en el núcleo de neuronas y células gliales1. En el momento actual, se distinguen tres grandes grupos según el tipo de proteína identificada en las inclusiones celulares: la DLFT con inclusiones de la proteína asociada al microtúbulo, tau (DLFT-tau), la DLFT con inclusiones compuestas por la proteína fijadora del ADN TAR 43 (DLFT-TDP) y la DLFT asociada a la proteína fusionada en el sarcoma (DLFT-FUS)2-3. De los casos con un diagnóstico histológico de DLFT, casi la mitad son una DLFT-tau, el 40% una DLFT-TDP, y el 5-10% restante, presentan una DLFT-FUS2,4. En torno a un 2 % de los pacientes quedan fuera de esta clasificación, ya que no forman parte de ninguno de estos tres grupos (DLFT-otros)3. Genéticamente es un grupo también heterogéneo; se han identificado numerosos genes causantes, de entre los cuales los más importantes son los que codifican para proteína tau asociada a los microtúbulos (MAPT) y progranulina (GRN)5, y la expansión del cromosoma 9 (C9ORF72), como causa más frecuente de DFT asociada a esclerosis lateral amiotrófica, tanto esporádica como familiar.
Estos grupos neuropatológicos engloban síndromes clínicos diversos6 que incluyen: las tres variantes clínicas de la demencia frontotemporal (DFT), la variante conductual de la DFT (vcDFT), la afasia progresiva no fluente (APNF) y la demencia semántica (DS), así como el síndrome de la parálisis supranuclear progresiva (SPSP) y el síndrome corticobasal (SCB). En algunos casos los pacientes con DFT presentan también características clínicas de enfermedad de motoneurona (EMN); en estos casos el diagnóstico sindrómico es el de DFT-EMN.
La DFT se caracteriza principalmente por la presencia de alteraciones cognitivas (disfunción ejecutiva y/o del lenguaje) y conductuales, mientras que el SCB y el SPSP se caracterizan por la presencia de un parkinsonismo atípico. No obstante, se observan casos en los que existe un solapamiento entre las distintas entidades mencionadas, dificultando el diagnóstico en las fases iniciales de la enfermedad. Algunos autores han llegado a postular que existe un continuum entre estos síndromes clínicos caracterizados por la presencia de deterioro cognitivo y parkinsonismo7. La investigación de tests que definan el patrón neuropsicológico y permitan un diagnóstico precoz ha mostrado cómo los dominios más afectados en la DLFT frente a la EA son el lenguaje y la atención8. No obstante es crítica la búsqueda de tests cuyo resultado ayude en la caracterización de las diferentes entidades de la DLFT en estadios iniciales. El objetivo de este artículo es revisar el patrón neuropsicológico de cada una de las entidades clínicas que se engloban dentro del concepto de la DLFT a partir de la literatura publicada más relevante sobre esta materia y dilucidar cuáles son las características neuropsicológicas que permiten diferenciar las distintas entidades entre sí y con la EA.
El objetivo de nuestro trabajo fue el de seleccionar trabajos bien diseñados, con diferentes grupos de pacientes definidos según criterios diagnósticos, sin límite de edad, duración de la enfermedad ni del tratamiento, en los que se hubieran llevado a cabo tests neuropsicológicos para el estudio de diferentes dominios cognitivos.
Identificación y selección de los estudios
Se ha llevado a cabo una búsqueda sistemática de los estudios publicados hasta mayo de 2014 en inglés o español. La búsqueda se realizó incluyendo citas bibliográficas indexadas en la base de datos de MEDLINE, en revistas relevantes, seleccionando artículos referenciados con los siguientes términos: demencia frontotemporal, degeneración lobar frontotemporal, perfil neuropsicológico, parkinsonismo y comportamiento.
Dos autores diferentes llevaron a cabo la búsqueda, eliminando publicaciones seleccionadas por duplicado y escogiendo estudios relevantes en base al resumen de los trabajos. Se obtuvo un total de 45 trabajos. En todos los trabajos se revisó el texto completo para la consulta de tablas y detalles de los estudios.
Inicialmente se llevó a cabo una recogida de los datos relevantes de cada trabajo, incluyendo el número de pacientes de cada grupo, número de grupos, test neuropsicológicos empleados, rediseño del estudio, puntuaciones obtenidas y comparaciones entre grupos realizadas. A partir de dicha tabla se recogieron los resultados finales descritos en cada trabajo y se llevó a cabo la revisión que a continuación se desarrolla, teniendo en cuenta los diferentes grupos de pacientes (Degeneración lobar frontotemporal y sus variantes, enfermedad de Alzheimer y controles) y separándolos por dominios cognitivos.
Variante conductual de la demencia frontotemporal
La variante conductual de la DFT (vcDFT) constituye al menos la mitad de los casos de DFT. Se caracteriza por una pérdida progresiva de habilidades interpersonales y/o de las funciones ejecutivas, con pérdida de la capacidad de respuesta emocional y aparición de alteraciones de comportamiento que incluyen desinhibición, obsesiones, estereotipias y alteraciones en el patrón de alimentación. La amnesia severa y precoz y la desorientación espacial son criterios de exclusión de esta entidad9.
La vcDFT es histopatológicamente heterogénea. Aproximadamente, la mitad de los pacientes tienen una histopatología tipo DLFT-tau, mientras que la otra mitad tienen una DLFT-TDP10. Por otro lado, los pacientes con una vcDFT de inicio muy temprano se pueden asociar a DLFT-FUS11. Los casos de vcDFT presentan una atrofia de la corteza frontal medial anterior, así como de la ínsula, cíngulo anterior y lóbulo temporal anterior12-13. Las pruebas de neuroimagen funcional como la tomografía de emisión de positrones con 18F-desoxiglucosa (PET-FDG) muestran un hipometabolismo de la corteza frontal medial, seguido de la frontolateral y la corteza temporal anterior, con una progresión antero-posterior12,14. Estos hallazgos de neuroimagen se han incorporado recientemente a los criterios diagnósticos de la DFT9.
En la valoración de la capacidad cognitiva global con pruebas como la Mattis Dementia Rating Scale (MDRS) o el test del reloj15, los pacientes con vcDFT obtienen mejores resultados que los pacientes con EA.
Memoria
En los tests que valoran la memoria episódica verbal y visual, los pacientes con vcDFT obtienen mejores puntuaciones que los sujetos con EA15-18. No obstante, en algunos trabajos, hasta un 10 % de los pacientes con una vcDFT confirmada mediante necropsia tienen afectación de la memoria episódica en estadios iniciales10,19-20, evidenciándose así que puede existir un solapamiento del patrón cognitivo de la EA y la DFT en determinados casos21.
Función ejecutiva
Estudios que comparan la vcDFT, DS, EA y controles sanos muestran una mayor afectación de la función ejecutiva y la atención en la vcDFT, frente a la mayor afectación de memoria episódica en EA18,22. No obstante, no todos los estudios muestran unanimidad en cuanto a la mayor afectación de la función ejecutiva en los pacientes con la vcDFT. De hecho, Giovagnoli y col. no encontraron diferencias significativas en la función ejecutiva entre un grupo de pacientes con vcDFT y otro con EA, utilizando pruebas como la Torre del Londres, el test de Weigl o la parte B del Trail Making Test (TMT)17. Estas discrepancias podrían deberse al pequeño tamaño muestral o las diferencias en los tests utilizados.
Recientemente, se ha publicado un estudio que compara el patrón de afectación frontal de pacientes con vcDFT y PSP, sin encontrar diferencias significativas. Estos autores23, no obstante, abogan por un mecanismo de afectación frontal diferente en ambos casos, aun compartiendo un patrón neuropsicológico, siendo las lesiones más extensas y resultado del daño de circuitos córtico-subcorticales en el caso de la PSP.
Función visuoespacial
Las habilidades visuoespaciales, medidas mediante tests como la copia de una figura o el ensamblaje de bloques, están conservadas en la vcDFT hasta los estadios más avanzados15, a diferencia de los pacientes con EA. De manera que si se compara la afectación de dominios frontales-ejecutivos, medida por la Frontal Assesment Battery (FAB), frente a la afectación posterior-perceptual, medida por la Perceptual Assesment Battery (PAB) -compuesta por secciones de integración visual, figuras y habilidades espaciales-, se observa que en los pacientes con la vcDFT predomina la afectación frontal y en pacientes con EA, predomina la afectación posterior24.
Praxias
Es interesante el trabajo de Giovagnoli y col17 en el que los pacientes con EA presentan mayor dificultad en la realización de praxias constructivas, comparados con la vcDFT, resultado ya observado por el grupo de Perri y col25, en el que los pacientes con la vcDFT realizaron mejor la copia de la figura de Rey y, concluyen que es este test, junto con la fluencia verbal, el que consigue discriminar de manera más fiable entre la EA y la vcDFT.
Lenguaje
En cuanto al lenguaje, hay datos contradictorios en los estudios publicados. Rascovsky y col objetivan que los pacientes con una vcDFT obtienen peores puntuaciones en fluencia verbal fonética que los pacientes con EA15, mientras que Giovagnoli y col no encuentran diferencias entre ambas entidades17. De nuevo estas diferencias podrían deberse a la diferencia en el tamaño muestral, la diferencia en los tests utilizados o a la realización de un análisis retrospectivo.
En este sentido, Thompson y col26 encontraron que los errores en pacientes con vcDFT fueron consecuencia de la perseverancia, la confabulación y de una menor organización.
Conducta
Los síntomas conductuales son un rasgo característico de la DFT-vc y es por esto que en numerosos trabajos se han utilizado escalas de comportamiento, como el NPI, para estudiar la prevalencia de estos síntomas en pacientes con DFT-vc frente a EA. Las alteraciones conductuales son más prevalentes en el grupo de pacientes con vcDFT y entre ellas, la desinhibición, euforia y la apatía son las más frecuentes25.
Demencia semántica
La DS constituye aproximadamente un 25-33% de los casos de DFT. Se caracteriza clínicamente por una alteración progresiva de la memoria semántica, que afecta inicialmente al conocimiento de las palabras. Los pacientes presentan un lenguaje fluido con evidente anomia, empobrecimiento del vocabulario y dificultad para la comprensión de las palabras27. La información visual y otros dominios no verbales se afectan tardíamente, apareciendo prosopagnosia y agnosia visual. Las alteraciones conductuales son sutiles e incluyen depresión, desinhibición, apatía y/o irritabilidad. Los cambios en la conducta alimentaria son frecuentes, sobre todo con apetencia por lo dulce o restricción de los gustos culinarios. Hasta el 75 % de los casos con una DS se asocian a una DLFT-TDP28, y el resto, a una DLFT-tau. Los pacientes con una DS muestran en la neuroimagen estructural una atrofia temporal antero-inferior asimétrica29, con mayor frecuencia izquierda (aproximadamente tres veces más frecuente). En casos en los que predomina una atrofia del lóbulo temporal derecho sobre el izquierdo, la presentación clínica se caracteriza por la presencia de prosopagnosia y alteraciones del comportamiento (denominándose variante temporal derecha de la DS). Los estudios de neuroimagen con PET-FDG que comparan pacientes con DS frente a controles sanos muestran un hipometabolismo temporal anterior de predominio izquierdo, y en la región orbitofrontal de manera bilateral30-31.
Como se ha mencionado previamente, una de las características clínicas fundamentales de la DS es la anomia, que se pone de manifiesto por la falta de palabras que son reemplazadas por términos más generales, en un discurso gramaticalmente correcto y adecuadamente pronunciado. El conocimiento semántico es pobre28. La comprensión parece no estar afectada, salvo que existan palabras complejas, las cuales pueden repetir pero no definir. Precisamente, estos fueron los hallazgos fundamentales descritos por Hodges y col32 al estudiar esta entidad en 5 pacientes. Posteriormente,33 este mismo autor comparó pacientes con DS, EA, vcDFT y controles, definiendo un perfil neuropsicológico para cada grupo, de manera que los pacientes con DS mostraban un deterioro marcado de la memoria semántica y anomia frente al grupo de EA y vcDFT. Estudios posteriores que también han comparado el rendimiento cognitivo de los pacientes con DS frente a EA, vcDFT y sujetos control han obtenido resultados similares18,24. El rendimiento cognitivo de pacientes con DS ha sido comparado en otros trabajos frente a sujetos con EA y otras entidades del espectro de la DLFT, como la DCB y la APNF. Nuevamente, los pacientes con DS mostraban una mayor afectación de la memoria semántica, la denominación, la definición de palabras y la fluencia verbal con respecto a estos grupos de pacientes16.
Afasia progresiva no fluente
La APNF se caracteriza por una pérdida progresiva del lenguaje. Más específicamente, los pacientes muestran un discurso poco fluido, agramatismo y un lenguaje telegráfico con parafasias fonéticas27. En algunos pacientes, acompañando a la afasia, se puede observar una apraxia del habla. Este trastorno se define como una alteración en la programación y en la planificación motora del habla, que da lugar a una incapacidad para llevar a cabo los movimientos necesarios para elaborar el discurso, en ausencia de dificultad para realizar movimientos orales no necesarios para el mismo. Con la evolución de la enfermedad se pueden encontrar signos parkinsonianos como rigidez y bradicinesia34-35, así como disartria, distonía o fenómeno del miembro ajeno. El 70 % de los pacientes con una APNF muestran una DLFT-tau36, sobre todo en los casos con apraxia del habla. Aunque menos frecuente, hay casos asociados a DLFT-TDP y hasta en un 30 % se puede encontrar patología tipo Alzheimer. Los estudios de neuroimagen estructural muestran una atrofia cerebral afectando fundamentalmente a la región posterior fronto-insular izquierda12, área en la que se observa un hipometabolismo en los estudios con PET-FDG.
Los pacientes con APNF obtienen peores puntuaciones que los sujetos con EA en pruebas de memoria de trabajo y de control ejecutivo, como se demuestra en test de fluencia verbal y repetición de dígitos, mientras que su rendimiento es mejor en pruebas de memoria episódica y tareas visuoespaciales16,37.
Síndrome de la parálisis supranuclear progresiva
El término SPSP se reserva actualmente para el diagnóstico clínico de los pacientes que presentan una sintomatología caracterizada por inestabilidad postural temprana con parálisis supranuclear de la mirada vertical, disartria y disfagia pseudobulbares, un síndrome rígido-acinético con escasa respuesta a la levodopa y disfunción frontal grave38. Por otro lado, el término PSP se reserva para el diagnóstico anatomopatológico, caracterizado por pérdida neuronal, gliosis y la presencia de ovillos neurofibrilares formados por agregados de tau e inclusiones oligodendrogliales, con afectación predominante de los ganglios basales y el troncoencéfalo39. El grupo de Lees y Williams abogan por definir diferentes entidades clínicas para evitar términos como el de PSP-atípica. De manera que diferencian entre síndrome de Richardson, la forma clásica y PSP-parkinsonismo, PSP-acinesia pura y freezing de la marcha, PSP-síndrome corticobasal y PSP-afasia progresiva no fluente, según los síntomas que la acompañan. Existe una muy buena asociación clínico-patológica entre la DLFT-tau y el SPSP36, aunque en una minoría de casos de SPSP se ha descrito patología TDP-4340. En cuanto a la patología molecular, la PSP, al igual que la DCB, es una taupatía de la forma de 4 repeticiones (4R). La resonancia magnética (RM) de pacientes con un SPSP suele mostrar una atrofia bilateral de estructuras profundas como el tálamo, el hipocampo, el globo pálido y el troncoencéfalo. En la PET-FDG se observa un hipometabolismo de la corteza prefrontal, el estriado, el mesencéfalo y el tálamo41-42. En los pacientes con un SPSP y evidente alteración cognitiva, se ha observado un mayor grado de atrofia e hipometabolismo frontal en la RM y la PET-FDG, respectivamente. Recientemente se ha publicado el papel del PET 18F-FDDNP como método de cuantificación y localización in vivo de los agregados de tau en SPSP, presentes en áreas subcorticales (estriado, tálamo, subtálamo, troncoencéfalo y sustancia blanca cerebelosa)43.
Las alteraciones conductuales que con más frecuencia se observan en pacientes con SPSP, al igual que en sujetos con SCB o enfermedad de Parkinson (EP), son la depresión y la ansiedad, seguidos por la apatía y las alteraciones del sueño44. Yatabe y col45 compararon los síntomas neuropsiquiátricos de pacientes con SPSP y DFT sin observar diferencias significativas en las puntuaciones totales o en las subescalas del Inventario Neuropsiquiátrico (NPI) entre ambos grupos de pacientes. Ambos grupos obtuvieron un perfil similar en apatía, comportamiento, desinhibición y en comportamiento antisocial. No obstante, las puntuaciones fueron significativamente más bajas en pacientes con SPSP en el inventario de estereotipias total (SRI), especialmente en los ítems de comportamiento en ingesta y cocina.
Las características del déficit cognitivo del SPSP fueron descritas por Steele y col en 196446, quien encontró que estos pacientes presentaban alteraciones de memoria y recuerdo, cálculo, pensamiento abstracto, atención, comprensión y apraxia, asociados a confusión, cambios en el comportamiento y en la personalidad, que podían llegar a preceder al trastorno del movimiento. Posteriormente, Albert y col definieron la enfermedad como una demencia subcortical en ausencia de afasia, apraxia o agnosia47. Las alteraciones cognitivas de los pacientes con SPSP son similares a las que presentan los pacientes con disfunción frontal, con olvidos aunque con preservación de la memoria a corto plazo y la memoria implícita48. En la valoración cognitiva global, medida con tests como el MMSE, la Dementia Rating Scale o la Addenbrocke´s Cognitive Examination, los pacientes con SPSP puntúan mejor que los pacientes con EA49.
Memoria
Los pacientes con un SPSP, presentan una alteración leve en el recuerdo libre y en la memoria remota, mientras que el reconocimiento se encuentra preservado50. La alteración de la memoria a largo plazo mejora significativamente con claves y, por lo tanto, se debe a la dificultad para acceder a la información51.
Función ejecutiva
La afectación frontal en el SPSP es temprana52, como lo demuestran las alteraciones que se observan al utilizar el inventario de comportamiento frontal en pacientes con esta entidad clínica53. Aunque inicialmente los tests de atención y orientación son normales, otras tareas más complejas como planificación, abstracción o razonamiento están alteradas54. Se observan también alteraciones en el razonamiento no verbal, como las matrices de Raven55-56, o el test de semejanzas. Una característica fundamental del deterioro cognitivo del SPSP es el enlentecimiento en el procesamiento de la información, con el consiguiente aumento en la latencia de respuesta. Este enlentecimiento se correlaciona con el rendimiento en tests frontales como el Wisconsin, pero no con la bradicinesia característica de esta entidad clínica57-58. Los estudios que comparan sujetos con un SPSP con otras entidades que cursan con parkinsonismo como la EP o la atrofia multisistémica (AMS) muestran que los primeros rinden peor en tareas ejecutivas. En este sentido, Paviour y col59 observaron peores puntuaciones en los sujetos con SPSP frente a otros parkinsonismos utilizando la FAB; más aún, con la progresión de la enfermedad, la caída en los rendimientos en pruebas de función ejecutiva es significativamente mayor en el SPSP50.
Función visuoespacial
Los pacientes con SPSP, comparado con sujetos con EP o AMS, presentan rendimientos bajos en tareas visuoespaciales, utilizando pruebas de velocidad (búsqueda visual) o en el test de orientación de líneas del Benton55. Con la progresión de la enfermedad, se evidencia un empeoramiento significativamente mayor en este dominio área cognitiva en pacientes con SPSP con respecto a pacientes con EP55.
Lenguaje
La alteración del lenguaje que tienen los pacientes con SPSP, al igual que los pacientes con SCB, se caracteriza por un discurso limitado a frases cortas o palabras aisladas aunque gramaticalmente correcto y sin parafasias. La apraxia del habla, que se puede encontrar en pacientes con APNF, se ha descrito también como manifestación de enfermedades degenerativas como la DCB y la PSP60, en ausencia de afasia. En fluencia verbal los pacientes con SPSP tienen un importante deterioro, sobre todo con clave fonética más que con semántica59, siendo peor el rendimiento, comparado con el de la EP o la AMS55.
Síndrome corticobasal
El término síndrome corticobasal, al igual que en el anterior apartado con el SPSP, se reserva para el diagnóstico clínico de pacientes que presentan un síndrome rígido-bradicinético asimétrico que no mejora con la administración de levodopa, asociado a determinados signos corticales como apraxia de las extremidades, mioclonías reflejas o fenómeno de la mano ajeno, ipsilaterales al parkinsonismo. En estos sujetos también suelen aparecer otros trastornos del movimiento como distonía o temblor de acción, así como inestabilidad postural, signos de afectación de la vía piramidal o alteraciones oculomotoras. El SCB se asocia con frecuencia a la DLFT-tau, fundamentalmente a la DCB, aunque también existen casos con patología tipo PSP o tipo enfermedad de Pick36. No obstante, se han descrito casos con patología Alzheimer o con inclusiones TDP61-64. Los pacientes con SCB muestran una atrofia asimétrica frontal, sobre todo del área motora suplementaria y corteza premotora, asociada o no a atrofia del lóbulo parietal y con menor frecuencia temporal. El patrón característico en la PET-FDG es un hipometabolismo contralateral al lado afectado clínicamente, con afectación predominante de la región parieto-temporal, prefrontal, cíngulo, corteza motora, caudado y tálamo41. Por otra parte, el término degeneración corticobasal (DCB) se reserva para el diagnóstico anatomopatológico caracterizado por depósitos de proteína tau y cuya expresión clínica es variable. Hasta el momento los criterios clínicos más extendidos para el diagnóstico de la DCB eran los de la Movement Disorders Society65. Recientemente, se han publicado los nuevos criterios diagnósticos de la DCB en los que se distinguen dos categorías (probable y posible) y cuatro fenotipos clínicos asociados a la DCB (SCB, síndrome frontal comportamental-espacial, APNF y el SPSP) 66.
Las alteraciones cognitivo-conductuales son frecuentes en el SCB. La alteración del comportamiento de los pacientes con SCB es similar a la que se observa en los pacientes con la vcDFT. Litvan y col, utilizando el NPI,67 describieron sobre todo depresión, aunque también apatía, desinhibición, irritabilidad y ansiedad. La demencia como primera manifestación en pacientes con SCB ha sido descrita por varios autores68-69 y, aunque durante años se consideró un criterio de exclusión para el diagnóstico del SCB/DCB70, en el momento actual apoya el diagnóstico71. En fases avanzadas hasta un 25 % de los pacientes con SCB tienen demencia. La memoria episódica parece estar alterada en aquellos pacientes con patología tipo DCB en el hipocampo68 y probablemente se deba a alteraciones fronto-subcorticales que dificultan la codificación y el recuerdo. Estas dificultades en el recuerdo mejoran significativamente con claves semánticas, al igual que ocurre en pacientes con el SPSP72. No obstante, la memoria está menos afectada que las funciones frontales y el lenguaje.
Función ejecutiva
La alteración cognitiva fundamental es una disfunción ejecutiva, seguida por las alteraciones del lenguaje. Al menos el 50% de los casos descritos de SCB tienen una disfunción lobar frontal con alteraciones en el comportamiento o bajas puntuaciones en tests frontales73-74. Los pacientes con SCB obtienen bajas puntuaciones en pruebas como el TMT-B, test de Stroop, fluencia verbal fonética y semántica. La afectación en tareas ejecutivas del SCB es mayor que la que presentan pacientes con EA y similar a la del SPSP72.
Función visuoespacial
Las alteraciones visuoespaciales, medidas mediante la Addenbrooke´s Cognitive Examination, son características de los pacientes con el SCB, y ayudan a distinguirlo de otros parkinsonismos atípicos, como la AMS o la PSP, así como de los pacientes con EA49, ya que son los pacientes con SCB los que obtienen peores rendimientos. Las dificultades en la copia y el dibujo (apraxia visuo-constructiva), y en la escritura, son frecuentes en el SCB62,68 como resultado de la afectación parietal.
Lenguaje
En el lenguaje, los pacientes con SCB presentan inicialmente anomia y progresivamente desarrollan dificultades para la expresión del lenguaje, mientras que la comprensión está conservada75. La afasia aparece sobre todo en el curso de la enfermedad de los pacientes que debutan con síntomas cognitivos, aunque también tiene lugar en aquellos con inicio motor76, mientras que las puntuaciones en el rendimiento cognitivo global y en comportamiento son similares en ambos grupos de pacientes77. Además, en el estudio de McMonagle y col76 se observó que la evolución de la afasia en los pacientes con SCB de inicio cognitivo es comparable a la de pacientes con APNF, no así a la de pacientes con EA. En relación con la lateralidad, teniendo en cuenta que la asimetría es una característica inherente a la DCB, se ha estudiado la posible relación entre la afectación motora del hemicuerpo derecho y la alteración del lenguaje. En este sentido no se han encontrado diferencias en las puntuaciones de los test de afasia (WAB) entre pacientes con inicio motor hemicorporal derecho o izquierdo, ni entre pacientes con mayor atrofia hemisférica derecha o izquierda76, aunque sí se encontró una predominancia entre pacientes con afectación motora derecha y afasia. Esta incongruencia podría deberse a un sesgo de selección, ya que acuden con más frecuencia a consulta de demencia y por tanto se estudia con más detalle la afasia en aquellos pacientes con mayor afectación de hemisferio izquierdo. Mientras que aquellos con bradicinesia hemicorporal izquierda con más frecuencia consultan en unidades de trastornos del movimiento. La falta de correlación entre atrofia, acinesia y afasia, puede deberse a que aunque predomine la atrofia en un hemisferio, cierto grado de afectación del hemisferio contralateral puede ser suficiente para manifestarse clínicamente.
Consideraciones finales
La DLFT es una entidad heterogénea con una expresión fenotípica muy variable, que incluye síndromes clínicos que se pueden solapar. La valoración cognitiva es fundamental para orientar el diagnóstico de estos pacientes en estadios iniciales. De hecho, cada uno de los síndromes clínicos descritos presenta un patrón neuropsicológico característico. En el caso de los pacientes con una vcDFT predomina la alteración de la función ejecutiva24, con preservación de la memoria episódica, tanto en dominios verbales (p.e. listas de palabras) como no verbales (p.e. reproducción de una figura compleja). Es la afectación de la función ejecutiva y la menor afectación de la memoria el dato que con más precisión diferencia vcDFT y EA. En la DS predomina un discurso fluido con empobrecimiento del lenguaje y evidente anomia. A diferencia de los pacientes con EA existe una relativa preservación de la memoria episódica. La APNF se caracteriza por una afectación del procesamiento gramatical, que se manifiesta por la escasa fluidez del discurso, agramatismo y parafasias fonéticas. Es la mayor afectación de memoria de trabajo y función ejecutiva lo que permite diferenciar los pacientes con APNF frente a la EA. El patrón cognitivo del SPSP se caracteriza por una afectación de la función ejecutiva desde estadios iniciales, en combinación con un enlentecimiento en el procesamiento de la información; mientras que la afectación de la memoria es leve y mejora con claves semánticas. En el SCB se afecta la función ejecutiva y el lenguaje, así como la función visuoespacial, fundamentalmente la copia y el dibujo, siendo este último aspecto importante en la distinción con otros parkinsonismos como el SPSP. De igual modo que ocurría en el SPSP existe una afectación del recuerdo, que mejora con claves, fundamentalmente semánticas.
En cuanto a la diferenciación entre DLFT y EA, los tests que discriminan entre ambas entidades y por tanto podrían predecir la progresión en estadios iniciales, son los de lenguaje y memoria y, concretamente el test de denominación por confrontación visual de Boston, el recuerdo y fluencia verbal.
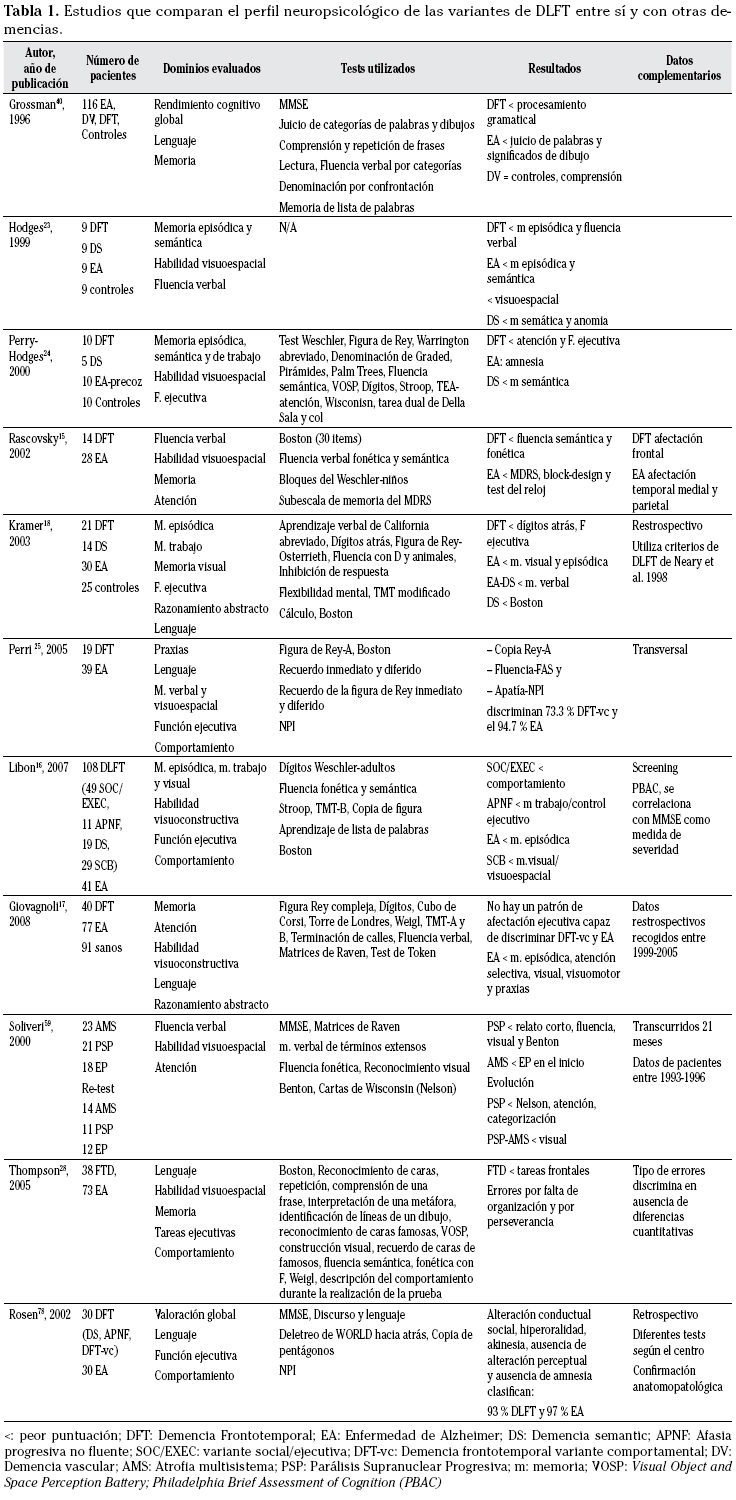
En resumen, el perfil neuropsicológico de la DLFT ha sido estudiado en cada una de las variantes y existen estudios que las comparan entre sí y frente a otras entidades, fundamentalmente la EA (Tabla 1). Sin embargo, es necesario llevar a cabo estudios longitudinales que permitan conocer cuál es la evolución y qué rasgos están presentes desde el inicio para poder distinguir las distintas entidades de manera precoz.
Bibliografía
1. Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, Cras P, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012; 124: 353-372. [ Links ]
2. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009; 117: 15-18. [ Links ]
3. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010; 119: 1-4. [ Links ]
4. Halliday G, Bigio EH, Cairns NJ, Neumann M, Mackenzie IR, Mann DM. Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol 2012; 124: 373-382. [ Links ]
5. Rohrer JD, Lashley T, Schott JM, Warren JE, Mead S, Isaacs AM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 2011; 134: 2565-2581. [ Links ]
6. Taipa R, Pinho J, Melo-Pires M. Clinico-pathological correlations of the most common neurodegenerative dementias. Front Neurol 2012; 3: 68. [ Links ]
7. Espay AJ, Litvan I. Parkinsonism and frontotemporal dementia: the clinical overlap. J Mol Neurosci 2011; 45: 343-349. [ Links ]
8. Yoshizawa H, Vonsattel JP, Honig LS. Presenting neuropsychological testing profile of autopsy-confirmed frontotemporal lobar degeneration. Dement Geriatr Cogn Disord 2013; 36: 279-289. [ Links ]
9. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134: 2456-2477. [ Links ]
10. Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004; 56: 399-406. [ Links ]
11. Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, et al. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 201; 122: 99-110. [ Links ]
12. Agosta F, Canu E, Sarro L, Comi G, Filippi M. Neuroimaging findings in frontotemporal lobar degeneration spectrum of disorders. Cortex 2012; 48: 389-413. [ Links ]
13. Pan PL, Song W, Yang J, Huang R, Chen K, Gong QY, et al. Gray matter atrophy in behavioral variant frontotemporal dementia: a meta-analysis of voxel-based morphometry studies. Dement Geriatr Cogn Disord 2012; 33: 141-148. [ Links ]
14. Franceschi M, Anchisi D, Pelati O, Zuffi M, Matarrese M, Moresco RM, et al. Glucose metabolism and serotonin receptors in the frontotemporal lobe degeneration. Ann Neurol 2005; 57: 216-225. [ Links ]
15. Rascovsky K, Salmon DP, Ho GJ, Galasko D, Peavy GM, Hansen LA, et al. Cognitive profiles differ in autopsy-confirmed frontotemporal dementia and AD. Neurology 2002; 58: 1801-1808. [ Links ]
16. Libon DJ, Xie SX, Moore P, Farmer J, Antani S, McCawley G, et al. Patterns of neuropsychological impairment in frontotemporal dementia. Neurolog 2007; 68: 369-375. [ Links ]
17. Giovagnoli AR, Erbetta A, Reati F, Bugiani O. Differential neuropsychological patterns of frontal variant frontotemporal dementia and Alzheimer´s disease in a study of diagnostic concordance. Neuropsychologia 2008; 46: 1495-1504. [ Links ]
18. Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK, et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 2003; 16: 211-218. [ Links ]
19. Caine D, Patterson K, Hodges JR, Heard R, Halliday G. Severe anterograde amnesia with extensive hippocampal degeneration in a case of rapidly progressive frontotemporal dementia. Neurocase 2001; 7: 57-64. [ Links ]
20. Graham A, Davies R, Xuereb J, Halliday G, Kril J, Creasey H, et al. Pathologically proven frontotemporal dementia presenting with severe amnesia. Brain. 2005; 128: 597-605. [ Links ]
21. Hornberger M, Piguet O. Episodic memory in frontotemporal dementia: a critical review. Brain 2012; 135: 678-692. [ Links ]
22. Perry RJ, Hodges JR. Differentiating frontal and temporal variant frontotemporal dementia from Alzheimer´s disease. Neurology 2000; 54: 2277-2284. [ Links ]
23. Lagarde J, Valabregue R, Corvol JC, Pineau F, Le Ber I, Vidailhet M, et al. Are frontal cognitive and atrophy patterns different in PSP and bvFTD? A comparative neuropsychological and VBM Study. PLoS One 2013; 8: e80353. [ Links ]
24. Mendez MF, McMurtray AM, Licht EA, Saul RE. Frontal-executive versus posterior-perceptual mental status deficits in early-onset dementias. Am J Alzheimers Dis Other Demen 2009; 24: 220-227. [ Links ]
25. Perri R, Koch G, Carlesimo GA, Serra L, Fadda L, Pasqualetti P, et al. Alzheimer´s disease and frontal variant of frontotemporal dementia - a very brief battery for cognitive and behavioural distinction. J Neurol 2005; 252: 1238-1244. [ Links ]
26. Thompson JC, Stopford CL, Snowden JS, Neary D. Qualitative neuropsychological performance characteristics in frontotemporal dementia and Alzheimer´s disease. J Neurol Neurosurg Psychiatry 2005; 7: 920-927. [ Links ]
27. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76: 1006-1014. [ Links ]
28. Hodges JR, Patterson K. Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol 2007; 6: 1004-1014. [ Links ]
29. Rohrer JD, Warren JD, Modat M, Ridgway GR, Douiri A, Rossor MN, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology 2009; 72: 1562-1569. [ Links ]
30. Diehl J, Grimmer T, Drzezga A, Riemenschneider M, Forstl H, Kurz A. Cerebral metabolic patterns at early stages of frontotemporal dementia and semantic dementia. A PET study. Neurobiol Aging 2004; 25: 1051-1056. [ Links ]
31. Desgranges B, Matuszewski V, Piolino P, Chetelat G, Mezenge F, Landeau B, et al. Anatomical and functional alterations in semantic dementia: a voxel-based MRI and PET study. Neurobiol Aging 2007; 28: 1904-1913. [ Links ]
32. Hodges JR, Patterson K, Oxbury S, Funnell E. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain 1992; 115: 1783-1806. [ Links ]
33. Hodges JR, Patterson K, Ward R, Garrard P, Bak T, Perry R, et al. The differentiation of semantic dementia and frontal lobe dementia (temporal and frontal variants of frontotemporal dementia) from early Alzheimer´s disease: a comparative neuropsychological study. Neuropsychology 1999; 13: 31-40. [ Links ]
34. Kertesz A, Martinez-Lage P, Davidson W, Munoz DG. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology 2000; 55: 1368-1375. [ Links ]
35. Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005; 128: 1996-2005. [ Links ]
36. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011; 122: 137-153. [ Links ]
37. Grossman M, Rhee J, Moore P. Sentence processing in frontotemporal dementia. Cortex 2005; 41: 764-777. [ Links ]
38. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurolog 1996; 47: 1-9. [ Links ]
39. Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010; 23: 394-400. [ Links ]
40. Yokota O, Davidson Y, Bigio EH, Ishizu H, Terada S, Arai T, et al. Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 2010; 120: 55-66. [ Links ]
41. Teune LK, Bartels AL, de Jong BM, Willemsen AT, Eshuis SA, de Vries JJ, et al. Typical cerebral metabolic patterns in neurodegenerative brain diseases. Mov Disord 2010; 25: 2395-2404. [ Links ]
42. Mascalchi M, Vella A, Ceravolo R. Movement disorders: role of imaging in diagnosis. J Magn Reson Imaging 2012; 35: 239-256. [ Links ]
43. Kepe V, Bordelon Y, Boxer A, Huang SC, Liu J, Thiede FC, et al. PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. J Alzheimers Dis 2013; 36:145-153. [ Links ]
44. Borroni B, Turla M, Bertasi V, Agosti C, Gilberti N, Padovani A. Cognitive and behavioral assessment in the early stages of neurodegenerative extrapyramidal syndromes. Arch Gerontol Geriatr 2008; 47: 53-61. [ Links ]
45. Yatabe Y, Hashimoto M, Kaneda K, Honda K, Ogawa Y, Yuuki S, et al. Neuropsychiatric symptoms of progressive supranuclear palsy in a dementia clinic. Psychogeriatrics 2011; 11: 54-59. [ Links ]
46. Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. A Heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 10: 333-359. [ Links ]
47. Albert ML, Feldman RG, Willis AL. The ´subcortical dementia´ of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 1974; 37: 121-130. [ Links ]
48. Litvan I. Cognitive disturbances in progressive supranuclear palsy. J Neural Transm Suppl 1994; 42: 69-78. [ Links ]
49. Bak TH, Rogers TT, Crawford LM, Hearn VC, Mathuranath PS, Hodges JR. Cognitive bedside assessment in atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2005; 76: 420-422. [ Links ]
50. Beatty WW, Scott JG, Wilson DA, Prince JR, Williamson DJ. Memory deficits in a demented patient with probable corticobasal degeneration. J Geriatr Psychiatry Neurol 1995; 8: 132-136. [ Links ]
51. Pillon B, Deweer B, Michon A, Malapani C, Agid Y, Dubois B. Are explicit memory disorders of progressive supranuclear palsy related to damage to striatofrontal circuits? Comparison with Alzheimer´s, Parkinson´s, and Huntington´s diseases. Neurology 1994; 44: 1264-1270. [ Links ]
52. Magherini A, Litvan I. Cognitive and behavioral aspects of PSP since Steele, Richardson and Olszewski´s description of PSP 40 years ago and Albert´s delineation of the subcortical dementia 30 years ago. Neurocase 2005; 11: 250-262. [ Links ]
53. Borroni B, Alberici A, Agosti C, Cosseddu M, Padovani A. Pattern of behavioral disturbances in corticobasal degeneration syndrome and progressive supranuclear palsy. Int Psychogeriatr 2009; 21: 463-468. [ Links ]
54. Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a frontal assessment battery at bedside. Neurology 2000; 12; 55: 1621-1626. [ Links ]
55. Soliveri P, Monza D, Paridi D, Carella F, Genitrini S, Testa D, et al. Neuropsychological follow up in patients with Parkinson´s disease, striatonigral degeneration-type multisystem atrophy, and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2000; 69: 313-318. [ Links ]
56. Soliveri P, Piacentini S, Girotti F. Limb apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology 2005; 64: 448-453. [ Links ]
57. Dubois B, Pillon B, Legault F, Agid Y, Lhermitte F. Slowing of cognitive processing in progressive supranuclear palsy. A comparison with Parkinson´s disease. Arch Neurol 1988; 45: 1194-1199. [ Links ]
58. Pirtosek Z, Jahanshahi M, Barrett G, Lees AJ. Attention and cognition in bradykinetic-rigid syndromes: an event-related potential study. Ann Neurol 2001; 50: 567-573. [ Links ]
59. Paviour DC, Winterburn D, Simmonds S, Burgess G, Wilkinson L, Fox NC, et al. Can the frontal assessment battery (FAB) differentiate bradykinetic rigid syndromes? Relation of the FAB to formal neuropsychological testing. Neurocase 2005; 11: 274-282. [ Links ]
60. Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS, Parisi JE, et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase 2005; 11: 283-296. [ Links ]
61. Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011; 70: 327-340. [ Links ]
62. Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff-Radford N, Caselli RJ, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999; 53: 795-800. [ Links ]
63. Chand P, Grafman J, Dickson D, Ishizawa K, Litvan I. Alzheimer´s disease presenting as corticobasal syndrome. Mov Disord 2006; 21: 2018-2022. [ Links ]
64. Tartaglia MC, Sidhu M, Laluz V, Racine C, Rabinovici GD, Creighton K, et al. Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol 2010; 119: 365-374. [ Links ]
65. Litvan I, Bhatia KP, Burn DJ, Goetz CG, Lang AE, McKeith I, et al. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18: 467-486. [ Links ]
66. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80: 496-503. [ Links ]
67. Litvan I, Cummings JL, Mega M. Neuropsychiatric features of corticobasal degeneration. J Neurol Neurosurg Psychiatry 1998; 65: 717-721. [ Links ]
68. Bergeron C, Pollanen MS, Weyer L, Black SE, Lang AE. Unusual clinical presentations of cortical-basal ganglionic degeneration. Ann Neurol 1996; 40: 893-900. [ Links ]
69. Grimes DA, Lang AE, Bergeron CB. Dementia as the most common presentation of cortical-basal ganglionic degeneration. Neurology 1999; 53: 1969-1974. [ Links ]
70. Lang AE, Bergeron C, Pollanen MS, Ashby P. Parietal Pick´s disease mimicking cortical-basal ganglionic degeneration. Neurology 1994; 44: 1436-1440. [ Links ]
71. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003; 54S: 15-19. [ Links ]
72. Pillon B, Blin J, Vidailhet M, Deweer B, Sirigu A, Dubois B, et al. The neuropsychological pattern of corticobasal degeneration: comparison with progressive supranuclear palsy and Alzheimer´s disease. Neurology 1995; 45: 1477-1483. [ Links ]
73. Wenning GK, Litvan I, Jankovic J, Granata R, Mangone CA, McKee A, et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry 1998; 64: 184-189. [ Links ]
74. Frisoni GB, Pizzolato G, Zanetti O, Bianchetti A, Chierichetti F, Trabucchi M. Corticobasal degeneration: neuropsychological assessment and dopamine D2 receptor SPECT analysis. Eur Neurol 1995; 35: 50-54. [ Links ]
75. Grossman M, D´Esposito M, Hughes E, Onishi K, Biassou N, White-Devine T, et al. Language comprehension profiles in Alzheimer´s disease, multi-infarct dementia, and frontotemporal degeneration. Neurology 1996; 47: 183-189. [ Links ]
76. McMonagle P, Blair M, Kertesz A. Corticobasal degeneration and progressive aphasia. Neurology 2006; 67: 1444-1451. [ Links ]
77. Kertesz A. Corticobasal degeneration. J Neurol Neurosurg Psychiatry 2000; 68: 275. [ Links ]
78. Rosen HJ, Hartikainen KM, Jagust W, Kramer JH, Reed BR, Cummings JL, et al. Utility of clinical criteria in differentiating frontotemporal lobar degeneration (FTLD) from AD. Neurology 2002; 58: 1608-1615. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
María del Mar Carmona-Abellán
Clínica Universidad de Navarra
Departamento de Neurología
Avenida Pío XII, 36.
31008 Pamplona. Navarra
E-mail:
mcabellan@unav.es
Recepción: 20 de agosto de 2014
Aceptación provisional: 22 de octubre de 2014
Aceptación definitiva: 3 de noviembre de 2014

