










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRCOE
Print version ISSN 1138-123X
RCOE vol.9 n.4 Jun./Aug. 2004
| Enfermedades ampollares
|
|

Bullous diseases of the oral cavity: Pemphigus
Jiménez-Soriano, Yolanda*
Díaz-Fernández, José Mª **
* Profesora asociada de Medicina Bucal.
Facultad de Medicina y Odontología de Valencia.
Médico Adjunto del Servicio de Estomatología
Hospital General Universitario de Valencia.
** Médico Adjunto del Servicio de Estomatología
Hospital General Universitario de Valencia.
Correspondencia
Yolanda Jiménez Soriano
C/ Salamanca 68, 10
46005 Valencia
E-mail: quin@ctv.es
Resumen: Dentro de las patologías que cursan con la formación de ampollas en la mucosa oral existe un grupo importante de enfermedades mucocutáneas entre las que se encuentra el pénfigo.
En este artículo pretendemos realizar una revisión y puesta al día de esta enfermedad centrándonos en sus dos variantes que se expresan fundamentalmente con manifestaciones orales: el pénfigo vulgar y el pénfigo paraneoplásico.
Revisamos los criterios etiopatológicos que nos llevan a comprender las manifestaciones clínicas, analizamos los puntos en que basamos el diagnóstico y los tratamientos que con mayor frecuencia se están utilizando en estos pacientes.
Es importante su conocimiento ya que son enfermedades que suelen iniciarse en la cavidad oral antes de expresarse en el resto del organismo, mejorando mucho el pronóstico de estos pacientes un diagnóstico y tratamiento temprano.
Palabras clave: Pénfigo vulgar, Pénfigo paraneoplásico.
Abstract: Ampullar lesions of the oral mucosa comprise an important group of mucocutaneous disorders – including pemphigus.
A review and update of this disease is provided, focusing on the two variants that fundamentally manifest in the oral cavity: pemphigus vulgaris and paraneoplastic pemphigus.
A review is provided of the etiopathogenic criteria underlying the clinical manifestations of the disease, with an analysis of the diagnosis and treatment modalities most commonly employed in such patients.
Knowledge of these aspects is important, since these disorders tend to develop in the oral cavity before extending to the rest of the body, and can contribute to ensure early diagnosis and treatment, with an improved prognosis.
Key words: Pemphigus, Paraneoplastic pemphigus
BIBLID [1138-123X (2004)9:4; julio-agosto 361-476]
Jiménez-Soriano Y, Díaz-Fernández JM. Enfermedades ampollares en la cavidad oral: pénfigo. RCOE 2004;9(4):439-447.
Introducción
Las enfermedades vesículo-ampollares que afectan a la mucosa oral constituyen un grupo importante de alteraciones por su dificultad diagnóstica, ya que a la exploración podemos apreciar úlceras o erosiones inespecíficas, por la fragilidad de las ampollas al traumatismo masticatorio y sobre todo por la gravedad y cronicidad que pueden alcanzar de alguna de estas enfermedades como el pénfigo, siendo fundamental la identificación de las lesiones así como el conocimiento de las enfermedades para orientar el diagnóstico e iniciar un correcto tratamiento precoz que mejora el pronóstico de estos pacientes1*.
El pénfigo es una enfermedad autoinmune, organoespecífica en la que se producen vesículas y ampollas en la piel y las mucosas por la acción de autoanticuerpos contra proteínas específicas localizadas en las uniones de las células del epitelio2,3.
Hay dos formas fundamentales de pénfigo que son el pénfigo vulgar (PV) con una participación importante de las mucosas y el pénfigo foliáceo (PF) que se expresa en la piel y sus respectivas variantes (pénfigo vegetante y pénfigo eritematoso); estos dos grupos se diferencian por sus características clínicas, los autoanticuerpos, y la histología. Además han sido descritas nuevas entidades mucho más raras como son el pénfigo paraneoplásico (PPN), IgApénfigo y pénfigo herpetiforme4. Las manifestaciones orales son muy comunes en el pénfigo vulgar y en el pénfigo paraneoplásico, siendo muy infrecuentes en los otros tipos de pénfigo por esto comentaremos fundamentalmente estas dos entidades clínicas5.
Epidemilogía
El pénfigo es una enfermedad poco frecuente con una incidencia del 0,5-3,2 casos por 100.000 personas por año en el mundo. El pénfigo vulgar es la variante más frecuente constituyendo el 70% de todos los casos de pénfigo. Afecta a todas las razas con una mayor incidencia en los judíos Ashkenazim y en los japoneses3.
La mayor prevalencia la observamos entre la cuarta y la sexta décadas de la vida, y la afectación juvenil es rara pero también ha sido descrita, siendo casos muy severos. Presenta una incidencia similar en ambos sexos5,6.
Etiopatogenia
Es probable que los factores genéticos puedan jugar un papel en la susceptibilidad a padecer la enfermedad, como lo confirman los casos de pénfigo descritos en familias, la mayor frecuencia de la enfermedad en determinados grupos étnicos como judíos y japoneses, o la asociación a genes del HLA. Genes del HLA clase I (HLA-A10, HLA- A26) y sobre todo la asociación con moléculas de HLA clase II (DR4, DR14)7**.
Parece que los factores genéticos no son exclusivos en la etiología de la enfermedad como lo hacen sospechar la detección de estos auto-Ac en pacientes sanos, o la baja tasa de concordancia entre gemelos monocigóticos. De alguna manera existe también la participación de factores ambientales y se han descrito fármacos, virus, alimentos, quemaduras, exposición-UV relacionándose con el pénfigo más que demostrándose su papel etiológico7**.
El pénfigo también se ha descrito asociado a otras enfermedades autoinmunes como artritis reumatoide, miastenia gravis, lupus eritematoso o anemia perniciosa.
Como hemos señalado antes es una enfermedad autoinmune órganoespecífica donde se producen autoanticuerpos contra antígenos localizados en los desmosomas de los queratinocitos, rompiendo la adhesión intercelular, llevando a la acantolisis y a la formación de ampollas intraepiteliales. Estos autoanticuerpos son fundamentalmente IgG, sobre todo IgG1, IgG4, IgG2 y IgG3, y con mucha menor frecuencia IgM.
Aunque la patogenicidad del pénfigo esta caracterizada por la producción de estos autoanticuerpos es probable que la respuesta de la inmunidad celular también juegue un importante papel. La asociación con moléculas de HLA clase II (DR4, DR14) va a facilitar la presentación de péptidos derivados de la desmogleina a clones específicos de linfocitos T CD4+, que segregan citoquinas Th2 (IL4, IL6, IL10) mediando estas en la producción de anticuerpos patógenos por las células B7**.
El papel patógeno de estos autoanticuerpos (Auto-Ac) ha sido sospechado clínicamente por la correlación existente entre la actividad de la enfermedad y los títulos de estos en sangre, siendo la recaída más frecuente en pacientes con títulos persistentes y elevados. Otro argumento clínico son los casos de pénfigo descritos en neonatos de madres con enfermedad activa durante el parto. Estos casos regresan espontáneamente con la eliminación de los anticuerpos maternos. Son muy útiles para demostrar el papel patógeno de los Auto-Ac los modelos de experimentación tanto en animales como in vitro mediante proteínas purificadas7**.
Estos Auto-Ac van dirigidos contra unos antígenos epiteliales que se han identificado en el pénfigo vulgar fundamentalmente con las moléculas de adhesión desmogleinas 3 (glucoproteina de 130 KD) que pertenece a la familia de las cadherinas y están localizadas en las uniones del estrato espinoso, produciéndose la acantolisis en capas profundas del epitelio. En el pénfigo foliáceo estos antígenos se han identificado con las desmogleinas 1 (160KD) localizadas en la capa granular del epitelio, produciendo así acantolisis en capas más superficiales del epitelio. La causa por la que la tolerancia inmunológica se rompe contra estas proteínas permanece desconocida8,9.
Recientes hallazgos aportan argumentos a favor de una inhibición específica de la función adhesiva de la desmogleina 1 y 3 por los correspondientes anticuerpos (Ac) en los diferentes tipos de pénfigo, explicando la distinta localización de las lesiones observadas en el pénfigo foliáceo, pénfigo vulgar mucoso, y pénfigo vulgar con participación mucocutánea. La Dsg3 es la responsable mayor de la adhesión de las células en la mucosa oral. En la piel la Dsg1 y Dsg3 ambas participan en la adhesión de las células de las capas más basales de la epidermis, mientras la Dsg1 solo es responsable de la adhesión de las capas superficiales de la epidermis. Esto puede explicar la correlación entre la acantolisis superficial en piel observada en el pénfigo foliáceo como resultado de anticuerpos anti-Dsg1, lesiones mucosas en el PV por Ac anti-Dsg3, y lesiones cutaneomucosas en el PV como resultado de la presencia combinada de ambos tipos de Ac anti-Dsg3 y anti-Dsg17**. (fig. 1 y 2)
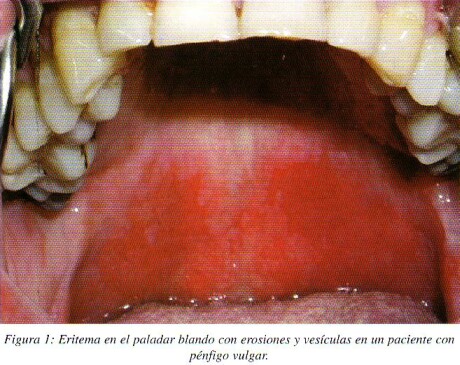

Clínica
El pénfigo vulgar se expresa principalmente con lesiones orales en un 88% de los pacientes, siendo además la mucosa oral el primer sitio donde se manifiesta en la mayoría de los casos (60%)2.
Generalmente comienza con lesiones muy inespecíficas, pasando meses desde el inicio hasta su diagnóstico. La mayoría de los pacientes presentan síntomas 2 a 6 meses antes de llegar al diagnóstico definitivo5.
La lesión elemental son las ampollas, que suelen ser múltiples, mal definidas, de distinto tamaño, de techo fino que se rompen fácilmente produciendo erosiones, superficiales, irregulares y muy dolorosas. El paciente presenta conjuntamente la formación de nuevas ampollas junto con otras ya evolucionadas y úlceras, expresando un carácter progresivo. Por lo tanto, podemos observar ampollas integras, ampollas que se están desprendiendo el techo de las mismas y que aparece como una auténtica membrana de tejido organizado que se puede separar con una sonda, o pseudomembranas que cubren erosiones que está formada fundamentalmente por un exudado inflamatorio y que no es un tejido organizado que pueda separarse con un explorador1*. (fig. 3)
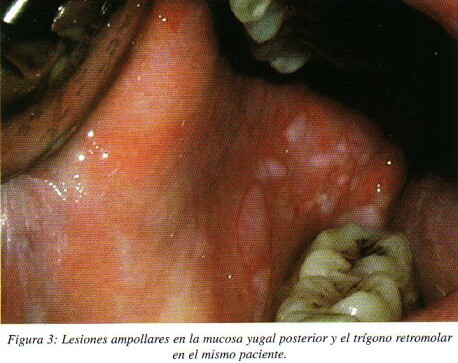
Cualquier localización de la mucosa oral puede estar afectada, pero con mayor frecuencia lo están las áreas de roce como es la mucosa bucal cerca del plano oclusal, los labios, la encía alveolar edéntula y el paladar blando. Las erosiones tienen tendencia a extenderse por toda la cavidad oral a partir de sus bordes, con un importante componente eritematoso3. En los labios las erosiones evolucionan a costras serohemáticas. Las lesiones curan sin dejar cicatrices. En estos pacientes el signo de Nikolsky es positivo ya que cualquier presión o traumatismo en la periferia de la lesión es capaz de incrementar rápidamente el tamaño inicial3,10.
En algunas ocasiones la participación de la encía se manifiesta como una gingivitis descamativa: encías eritematosas, brillantes, finas, con vesículas en la superficie que al rozarlas levemente se desprende el epitelio quedando expuesto el conectivo adyacente hemorrágico, expresando su carácter descamativo y erosivo11. (fig. 4)
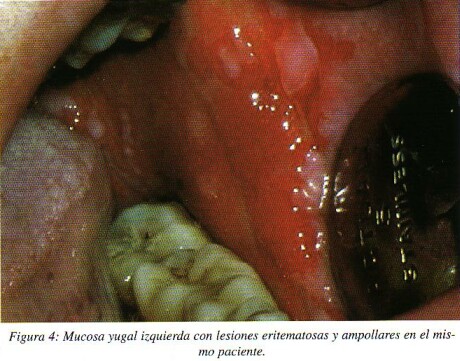
Cuando el pénfigo tiene manifestaciones en la piel, estas suelen aparecer tras meses o años después de las lesiones orales y se manifiestan como ampollas sobre una piel normal o ligeramente eritematosa, localizadas en cualquier parte del cuerpo pero con mayor frecuencia en cuero cabelludo, cara y parte superior del tronco10. Scully en su estudio determina que una cuarta parte de los pacientes con lesiones en la cavidad oral desarrollan en el transcurso del tiempo lesiones en la piel manifestándose meses o años tras las lesiones intraorales5.
También pueden afectarse otras mucosas como la faringe principalmente, la nariz, la laringe, el esófago, la uretra, la vulva, el cervix, el recto y la conjuntiva, aunque con mucha menor frecuencia y mayor gravedad2,5,6,12.
Pruebas complementarias
El diagnóstico clínico de sospecha tiene que ser confirmado por estudios complementarios que varían ampliamente en su especificidad y en su complejidad.
La citología exfoliativa tomada en fase de ampolla o vesícula muestra las células acantolíticas de Tzanck que son células epiteliales libres, redondeadas, de bordes desflecados y núcleos hipercromáticos, con citoplasma homogéneo eosinófilo en una tinción de Papanicolau; sin embargo estás células no son patognomónicas del PV y pueden observarse también en otras enfermedades ampollares13.(fig.5)
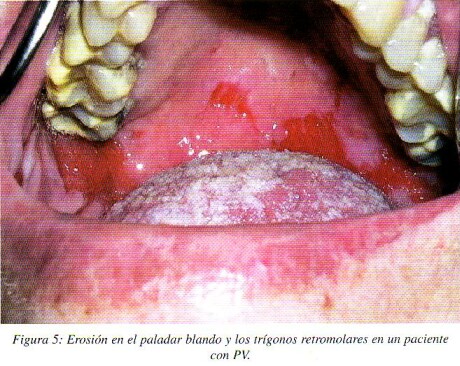
En el estudio histológico del PV observamos inicialmente un edema intercelular en los estratos suprabasales del espinoso con la formación de hendiduras y acantolisis, que llevan a la formación de ampollas.
Los estudios de inmunofluorescencia directa (IFD) muestran el marcaje en la superficie de las células epiteliales, indicando depósito de inmunoglobulinas en la membrana de los queratinocitos (espacio intercelular). Estos auto-anticuerpos depositados son principalmente IgG y mucho menos frecuente IgM. También puede detectarse depósito de complemento, especialmente la fracción C3, con la misma distribución7**.
La inmunofluorescencia indirecta (IFI) detecta anticuerpos circulantes anti-superficie de las células epiteliales (antisustancia intercelular) fundamentalmente IgG4 e IgG1, y menos frecuentemente la IgG3. Los títulos de auto-Ac a pénfigo suelen reflejar la actividad de la enfermedad, y son útiles para evaluar la evolución y el tratamiento. La recuperación se correlaciona con la disminución de los títulos de IgG4 permaneciendo la IgG1 positiva, demostrando el importante papel patógeno de la IgG47**.
Los estudios de inmunoprecipitación en la actualidad son considerados uno de los más definitivos e identifican los antígenos diana reconocidos por los autoanticuerpos del paciente, reconociéndose en el PV la Dsg 3 en el PF, la Dsg1 y en el PPN plakinas. Esta asociación facilita la descripción de la enfermedad; sin embargo algunos antígenos son reconocidos en diferentes tipos de pénfigo. Así la Dsg1 y la Dsg3 pueden ser detectadas en el suero de los pacientes con PV. Además este solapamiento de antígenos se ha demostrado relacionado con las manifestaciones clínicas del PV. El suero de pacientes de PV con manifestaciones clínicas exclusivas en mucosas solo contienen IgG anti-Dsg3 mientras que el suero de pacientes con manifestaciones mucocutáneas reaccionan contra la Dsg1 y la Dsg32,6,7**.
El diagnóstico de pénfigo se realiza por la sospecha clínica de las lesiones, y se tiene que confirmar por la presencia de acantolisis en la biopsia y por la determinación de anticuerpos bien en la sangre o en los tejidos.
Tratamiento
Los fármacos de elección para el tratamiento del pénfigo son los corticoides. Así lo refleja el cambió que sufrió la enfermedad con la introducción de los mismos disminuyendo drásticamente las tasas de mortalidad de estos pacientes. Sin embargo, las altas dosis y el tiempo prolongado de su utilización llevan a la aparición de graves efectos secundarios. Por esto la utilización de terapias adyuvantes con agentes inmunosupresores como la azatioprina, ciclofosfamida, metrotexate, ciclosporina, y cada vez se extiende más el empleo de nuevos agentes como el micofenolato mofetil o el tacrolimus como alternativas inmunosupresoras en monoterapia o terapia combinada con bajas dosis de corticoides14,15**,16.
El objetivo del tratamiento es reducir los anticuerpos que producen el daño tisular. En el momento actual no existe ninguna terapia que suprima específicamente los anticuerpos antidesmogleina, por lo que el tratamiento sigue basado en la inmunosupresión no específica.
El tratamiento inicial del pénfigo vulgar requiere el uso de corticoides sistémicos tales como la prednisona pudiendo comenzar con dosis de 1 mg/kg/día. Una vez controlado el brote hay que ir reduciendo progresivamente la dosis y pasando a régimen de administración a días alternos con el fin de minimizar los efectos secundarios, hasta llegar a alcanzar dosis mínimas de mantenimiento.
La vía tópica con corticoides se suele aplicar como apoyo a la administración por vía oral o como terapia de mantenimiento. Se puede administrar fluocinolona al 0,05% o propionato de clobetasol al 0,05%. Para las lesiones localizadas principalmente en encías se pueden administrar los corticoides en férulas oclusales17.
Si esta monoterapia con corticoides no controla las lesiones, no impide las recaídas o produce efectos secundarios importantes está indicada la terapia combinada. Los efectos adversos más frecuentes de los corticoides son la osteoporosis, el empeoramiento o desencadenamiento de diabetes y la hipertensión15**.
Generalmente la terapia combinada se inicia con la suma de inhibidores de la síntesis de la purina como el mofetil micofenolato o la azatioprina. El mofetil micofenolato parece desarrollar menos efectos secundarios presentando un mayor perfil de seguridad. Tiene un inicio de acción lento requiriendo a menudo de dos a tres meses para el control de los anticuerpos y es una terapia cara. La dosis recomendada es de 2 g/día. Los efectos secundarios más comunes son linfopenia, alteraciones gastrointestinales, intolerancia ocasional debido al dolor óseo y fatiga y aumento de la incidencia de infecciones. Necesita una monitorización sanguínea con un recuento hemático periódico. Este tratamiento, sin embargo, no siempre resulta efectivo en todos los pacientes con pénfigo14,15**,18,19.
La azatioprina ha mostrado ser más segura para alcanzar el control de la enfermedad y es más barata pero presenta una mayor incidencia de efectos Secundarios; estos incluyen leucopenia, pancitopenia, hepatotoxicidad, nauseas y fiebre. El riesgo de padecer estos efectos disminuye determinando la actividad del enzima tiopurina metil transferasa (TPMT). Este enzima metaboliza la azatioprina y según la actividad nos dan la dosis ajustada para cada paciente. Para pacientes con niveles normales de TPMT la dosis recomendada es de 3 mg/kg/día. La azatioprina presenta actividad antiinflamatoria con dosis de 2 mg/k/día pero esta no es eficaz para la inhibición de la producción de auto-Ac6,15**,16.
La ciclofosfamida es un agente alquilante utilizado de manera muy efectiva en el tratamiento del pénfigo pero que presenta gran toxicidad temprana como mielosupresión y cistitis hemorrágica y tardía aumentando el riesgo de padecer linfomas, leucemias y cáncer de vejiga. También está asociada con un mayor riesgo de esterilidad. La dosis recomendada es de 2-3 mg/kg/día junto con una intensa hidratación. Estos pacientes necesitan un seguimiento con recuento hemático y analítica urinaria semanalmente al principio y cada dos semanas posteriormente. Algunos autores han intentado disminuir su potencial tóxico administrándola en terapia pulse, que hace referencia a la infusión intravenosa discontinua de dosis muy altas de fármaco en un periodo corto de tiempo, se introdujo con el objeto de disminuir la acumulación del fármaco y así los efectos secundarios del mismo. Se han administrado regimenes de 100 mg de dexametasona en perfusión iv lenta durante tres días consecutivos, junto 500 mg de ciclofosfamida aplicada un solo día, esto se repite una vez al mes20. Este tratamiento se ha mostrado menos tóxico pero también menos efectivo16.
En pacientes con gran agresividad clínica o rápido deterioro del estado general se ha utilizado la plasmaféresis como terapia adyuvante junto con la administración de corticoides e inmunosupresores. La plasmaféresis es la terapia más directa para reducir la cantidad de anticuerpos patógenos en el suero. Esta reducción produce un efecto rebote en la síntesis de Ac por los linfocitos B debido a la eliminación del mecanismo de feedback; sin embargo esto se puede frenar con la administración de ciclofosfamida ya que produce una inhibición de la proliferación de linfocitos.
También se ha utilizado la administración de inmunoglobulinas intravenosas. Estas pueden provocar una rápida reducción de los autoanticuerpos de la circulación pero provocan un importante aumento en el catabolismo proteico y una mejoría a corto plazo no alargando los periodos de remisión.
La ausencia de estudios controlados a largo plazo para valorar la eficacia de los tratamientos ha llevado a la utilización de una amplia variedad de fármacos en el pénfigo. Entre los fármacos utilizados están el metotrexate, la dapsona, agentes antimaláricos, y terapia con oro (crisoterapia). Estos agentes no reducen la síntesis de Ac, por lo que su eficacia es discutida15**.
Evolución y pronóstico
Antes de la introducción de los corticoides, como hemos comentado anteriormente, la historia natural del pénfigo era hacia la extensión progresiva presentando una tasa de mortalidad a los dos años del 50% y del 100% a los cinco años. El uso de los corticoides redujo la tasa de mortalidad a menos del 5%; sin embargo está asociado a una importante tasa de morbilidad por su utilización prolongada16.
La eficacia del tratamiento se valora principalmente por la mejoría clínica y secundariamente por los parámetros de laboratorio con la disminución de los Ac séricos. Aunque no existen criterios estandarizados para valorar esta mejoría, los criterios clínicos que la determinan son la ausencia de nuevas lesiones, curación de las existentes y signo de Nikolsky negativo16.
El pénfigo puede evolucionar de manera variable hacia el control de la enfermedad sin requerir terapia de mantenimiento, hacia la necesidad de administrar una terapia de mantenimiento a dosis mínima (corticoides junto inmunosupresores), o incluso a la muerte del paciente5. En un estudio realizado por Scully de pacientes con pénfigo oral un 12% de ellos fueron controlados con corticoides tópicos, un 18% curó con corticoides sistémicos, un 76% requirió terapia de mantenimiento y un 6% de los pacientes murió5.
Hoy en día la mayor causa de mortalidad del pénfigo sigue siendo las complicaciones del tratamiento sistémico con los corticoides y los inmunosupresores, por lo que se necesita una cuidadosa monitorización de estos fármacos, siendo las infecciones la principal causa de mortalidad5.
Es importante resaltar el valor del diagnóstico y tratamiento precoz de estos pacientes ya que mejora el pronóstico y curso de la enfermedad6. También aquellos pacientes con una rápida respuesta al tratamiento tienen mejor pronóstico.
PENFIGO PARANEOPLÁSICO (PPN) aunque es mucho menos frecuente que el PV cabe una mención especial por la gran frecuencia y intensidad con que se manifiesta en la cavidad oral.
Fue descrito inicialmente por Anhalt y colaboradores en 1990, separándolo del pénfigo, como una enfermedad autoinmune causada por una alteración linfoproliferativa subyacente, con unos criterios diagnósticos clínicos, histológicos, e inmunológicos que lo definen21**.
1) clínicamente se caracterizan por una severa afectación oral (estomatitis) y lesiones polimorfas cutáneas que afectan preferentemente al tronco, extremidades, palmas y plantas en un paciente con una neoplasia oculta o confirmada.
2) histológicamente se evidencia un proceso cutáneo liquenoide, a menudo combinado con un despegamiento intraepitelial.
3) la IFD detecta IgG y complemento localizados en el espacio intercelular del epitelio junto con depósito lineal o granular del complemento en la zona de la membrana basal.
4) Autoanticuerpos séricos que se unen no solo a la superficie de las células del epitelio escamoso de piel y mucosas como en el pénfigo, sino también al epitelio simple, columnar y transicional, detectados por IFI y siguiendo el patrón típico del pénfigo (positiva para los Ac antisustancia intercelular).
5) los estudios de inmunoprecipitación reconocen la presencia de antígenos específicos de los desmosomas y hemidesmosomas: desmoplakina I (250KD), BPAg (230KD), envoplakin y desmoplakin II (210KD), periplakin (190KD) y un Ag indeterminado de 170 KD y la desmogleina 1 y 34,21**,22.
En 1993 Camisa y Helm revisaron los criterios diagnósticos y los divideron en mayores y menores. Los criterios mayores incluyen: una severa afectación oral y cutánea polimorfa, una neoplasia interna y anticuerpos determinados por inmunoprecipitación.
Los criterios menores incluyen el patrón histológico de acantolisis, IFD mostrando depósito de IgG marcando los espacios intercelulares y la zona de la membrana basal y los anticuerpos séricos detectados por IFI. Para el diagnóstico de PPN se requieren tres criterios mayores o dos menores y dos mayores4,22.
En aproximadamente dos tercios de los casos de PPN ocurren en pacientes donde ya se tenía conocimiento de una neoplasia, y en el tercio restante la lesión neoplásica fue detectada después de la enfermedad mucocutánea4.
Está asociado con mayor frecuencia a neoplasias hematológicas (84% de los casos) sobre todo linfomas no Hodgkin (42%), leucemias linfocitarias crónicas (29%), enfermedad de Castleman (10%) y otras. También se ha descrito asociado a neoplasias no hematológicas (16% de los casos)23.
Existe una participación de la mucosa oral importante en forma de mucositis dolorosa y persistente en todos los casos descritos y es el primer signo de la enfermedad en el 45% de los pacientes. En algunos pacientes solo existe afectación de la orofaringe sin llegar manifestarse en la piel21**. La estomatitis se manifiesta con erosiones y ulceraciones difusas severas, persistentes, muy dolorosas, y extremadamente resistentes al tratamiento y con un importante componente necrótico. Pueden afectar cualquier localización de la cavidad oral, sobre todo los bordes laterales de la lengua y característicamente existe también participación del borde rojo de los labios. También se observa una participación importante de la faringe aunque pueden aparecer lesiones en todas las mucosas del organismo presentando la mayoría de los pacientes una severa conjuntivitis pseudomembranosa4. Las lesiones cutáneas son variables y polimorfas observándose ampollas, erosiones y lesiones en diana, recordando en ocasiones un cuadro de eritema multiforme o necrosis epidérmica tóxica por su gravedad. Es característica la localización en palmas y plantas de los pies.
El PPN es el único tipo clínico de pénfigo que se manifiesta en tejidos que no están tapizados por epitelio escamoso estratificado desarrollando en aproximadamente un 30-40% de los casos lesiones pulmonares que pueden llevar a un fatal desenlace21**.
Las características histopatológicas del PPN son variables reflejando el polimorfismo de sus manifestaciones clínicas con acantolisis intraepitelial suprabasilar, necrosis de queratinocitos y degeneración vacuolar entre las más frecuentes. En la IFD se detecta depósitos intercelulares de IgG y C y además el depósito del complemento en la zona de la membrana basal que lo diferencia del PV. Robison alerta de que es más común observar falsos negativos en los resultados de inmunofluorescencia en el PPN que en el PV, de aquí la necesidad de tomar repetidas biopsias. La IFI utilizando un epitelio no estratificado (vejiga urinaria de rata) y un epitelio estratificado (esófago de mono) son utilizadas como sustrato para la IFI. El suero de pacientes con PV solo reacciona con el esófago de mono mientras el suero de pacientes con PPN reacciona con ambos. Sin embargo existen falsos negativos en un 25% de los pacientes necesitando realizar los estudios de inmunoprecipitación para confirmar el diagnóstico por identificación del complejo antigénico4.
El tratamiento de estos pacientes está condicionado en parte por la neoplasia asociada. Los pacientes con tumores resecables quirúrgicamente como tumores benignos, tumor de Castleman o timomas curan o mejoran ampliamente tras la resección del mismo. La curación completa es observada de 6 a 18 meses tras la escisión del tumor.
El PPN asociado a una neoplasia maligna generalmente no se resuelve a pesar del control o cura de la neoplasia, indicando que una vez que la enfermedad autoinmune se desencadena puede progresar de manera independiente. No hay evidencia de una pauta de tratamiento efectivo en estos pacientes, describiéndose que con corticoides orales a dosis de 0,5-1 mg/kg pueden mejorar las lesiones. Por esto en estos pacientes se han utilizado una multitud de tratamientos fallando en la mayoría de ellos incluyendo inmunosupresión con ciclofosfamida, azatioprina, oro, dapsona, plasmaféresis, inmunoglobulinas intravenosas a altas dosis, terapia pulse con ciclofosfamida20.
El fallecimiento de estos pacientes se ha relacionado con múltiples causas entre ellas sepsis, hemorragias gastrointestinales, fallo multiorgánico y fallo respiratorio21**.
Bibliografía recomendada
Para profundizar en la lectura de este tema, el/los autor/es considera/an interesantes los artículos que aparecen señalados del siguiente modo: *de interés **de especial interés.
1*. Bermejo-Fenoll A, López-Jornet P. Diagnóstico de las enfermedades vesiculares y ampollares de la mucosa bucal: desórdenes de la cohesión intraepitelial y de la unión epitelioconectiva. Med Oral 1996;1:24-43. [ Links ]
Es un buen artículo que realiza un excelente diagnóstico diferencial de las lesiones ampollares.
2. Weinberg MA, Insler MS, Campen RB. Mucocutaneous features of autoinmune blistering diseases. Oral Med Oral Pathol Oral Radiol Endod 1997;84:517-34. [ Links ]
3. Bagán JV. Enfermedades ampollares de la cavidad oral (I): pénfigos. En: Bagán JV, Ceballos A, Bermejo A, Aguirre JM, Peñarrocha M, eds. Medicina Oral. Barcelona:Masson,1995:220-6 [ Links ]
4. Robinson ND, Hashimoto T, Amagai M, Chan LS. The new phemphigus variants. J American Academy Dermatol 1999;40:649-71. [ Links ]
5. Scully, Paes de Almeida, Porter, Gilkes. Pemphigus vulgaris: the manifestations and long term management of 55 patients with oral lesions. British J Dermatol 1999;140:84-92 [ Links ]
6. Robinson JC, Lozada-Nur F. Oral pemphigus vulgaris. A review of the literature and a report on the management of 12 cases. Oral Med Oral Pathol Oral Radiol Endod 1997;84:349-55. [ Links ]
7**. Martell P, Joly P. Pemphigus: autoinmune diseases of keratinocytes adhesion molecules. Clinics in Dermatology 2001;19:662-74. [ Links ]
Los autores explican la patogenia del pénfigo de manera muy actualizada y profundizando paso a paso en los en los distintos factores que intervienen.
8. Lin MS, Swartz SJ, López A, Ding X, Fairley JA, Díaz LA. T–lymphocytes from a subset of patients with pemphigus vulgaris respond to both desmoglein 3 and desmoglein 1. J Invest Dermatol 1997;109:734-7 [ Links ]
9. Hertl M, Veldman C. T–cellular autoinmunity against desmogleins in pemphigus, an autoantibody-mediated bullous disorder of the skin. Autoinmun Rev 2003;2:278-83 [ Links ]
10. Milián MA, Jiménez Y. Enfermedades ampollares de la cavidad oral: pénfigo y penfigoide. RCOE 1997;2:687-95 [ Links ]
11. Castellanos JL. Enfermedades gingivales de origin immune. Med Oral 2002;7:271-83. [ Links ] [ Links ]
13. Mignona MD, Muzio L, Zeppa P, Ruocco V, Bucci E. Immunocytochemical detection of autoantibody deposits in Tzanck smears from patients with oral pemphigus. J Oral Pathol Med 1997;26:254-7. [ Links ]
14. Stanley JR. Therapy of pemphigus vulgaris. Arch Dermatol 1999;135:76-8. [ Links ]
15**. Mimouni D, Anhalt GJ. Pemphigus. Dermatologic therapy 2002;15:362-8. [ Links ]
Los autores realizan una revisión actualizada y amplia de los distintos tratamientos utilizados en pacientes con pénfigo.
16. Mimouni D, Nousari CH, Cummis DL, Kouba DJ, David M, Anhalt GJ. Differences and similarities among expert opinions on the diagnosis and treatment of pemphigus vulgaris. J Am Acad Dermatol 2003;49:1059-62. [ Links ]
17. Gonzalez-Moles MA, Morales P, Rodriguez-Archilla A, Ruiz-avila I, Gonzalez-Moles S. Treatment of severe chronic oral erosive lesions with clobetasol propionate in aqueous solution. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002;93:264-70. [ Links ]
18. Enk AH, Knop J. Mycophenolate is effective in the treatment of phemphigus vulgaris. Arch Dermathol 1999;135:54-6 [ Links ]
19. Powell AM, Albert S, Al fares S, Harman KE, Setterfield J, Bhogal B, Blank MM. An evaluation of the usefulness of mycophenolato mofetil in pemphigus. British J Dermatol 2003;149:138-45. [ Links ]
20. Pasricha JS, Siddhartha S. Curative effect of dexamethasone-cyclophosphamide pulse therapy for the treatment of phemphigus vulgaris. Inter J Dermatol 1992;31:875-7. [ Links ]
21**. Anhalt GJ. Paraneoplastic pemphigus. J Investigative Dermatology Symposium Proceedins 2004;9:29-33. [ Links ]
Es una buena revisión y puesta al día del pénfigo paraneoplásico.
22. Allen CM, Camisa C. Paraneoplastic pemphigus: a review of the literature. Oral Diseases 2000;6:208-14. [ Links ]
23. Kaplan I, Hodak E, Ackerman L, Mimouri D, Anhal GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncology 2004;40:553-62. [ Links ]

