










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkInternational Microbiology
versión impresa ISSN 1139-6709
INT. MICROBIOL. vol.8 no.4 dic. 2005
| RESEARCH ARTICLE | |||
|
| |||
| Genomic divergence of Escherichia coli strains: evidence for horizontal transfer and variation in mutation rates
Summary. This report describes the sequencing in the Escherichia coli B genome of 36 randomly chosen regions that are present in most or all of the fully sequenced E. coli genomes. The phylogenetic relationships among E. coli strains were examined, and evidence for the horizontal gene transfer and variation in mutation rates was determined. The overall phylogenetic tree indicated that E. coli B and K-12 are the most closely related strains, with E. coli O157:H7 being more distantly related, Shigella flexneri 2a even more, and E. coli CFT073 the most distant strain. Within the B, K-12, and O157:H7 clusters, several regions supported alternative topologies. While horizontal transfer may explain these phylogenetic incongruities, faster evolution at synonymous sites along the O157:H7 lineage was also identified. Further interpretation of these results is confounded by an association among genes showing more rapid evolution and results supporting horizontal transfer. Using genes supporting the B and K-12 clusters, an estimate of the genomic mutation rate from a long-term experiment with E. coli B, and an estimate of 200 generations per year, it was estimated that B and K-12 diverged several hundred thousand years ago, while O157:H7 split off from their common ancestor about 1.5-2 million years ago [Int Microbiol 2005 8(4):271-278] Key words: Escherichia coli strains · experimental evolution · evolution rates · horizontal gene transfer · molecular evolution | ||
| |||
Divergencia genómica de algunas cepas de Escherichia coli: pruebas de la transferencia horizontal y variación en las velocidades de mutación Resumen. Se secuenciaron 36 regiones del genoma de Escherichia coli B elegidas al azar y que están presentes en la mayoría o en todos los genomas de E. coli secuenciados. Se examinaron las relaciones filogenéticas entre cepas de E. coli y se buscaron pruebas de transferencia génica horizontal y de variación en la tasa de mutación. El árbol filogenético conjunto de genes indica que E. coli B y K-12 son las cepas con un parentesco más estrecho, mientras que E. coli O157:H7 se encuentra más alejada y aún más lo están Shigella flexneri 2a y E. coli CFT073, siendo esta última la más distante de todas. En el grupo B, K-12 y O157:H7, varias regiones indican que hay topologías alternativas. La transferencia génica horizontal es una explicación plausible de estas incongruencias filogenéticas, pero también hemos hallado pruebas de una evolución más rápida en sitios sinónimos en el linaje O157:H7. Así pues, una interpretación más profunda de estos resultados queda confundida por una asociación entre unos genes que muestran una evolución más rápida y otros que son transferidos horizontalmente. Usando genes que apoyan los grupos B y K-1, y empleando una estima de la tasa de mutación obtenida a partir de un experimento de evolución a largo plazo con E. coli B y suponiendo 200 generaciones por año, se estimó que las cepas B y K-12 divergieron hace varios cientos de miles de años, mientras que O157:H7 se separó de su ancestro común hace entre 1,5 y 2 millones de años [Int Microbiol 2005 8(4):271-278]. Palabras clave: cepas de Escherichia coli · evolución experimental · velocidad de evolución · transferencia horizontal de genes · evolución molecular | Divergência genômica de algumas cepas de Escherichia coli: provas da transferência horizontal e variação na velocidades de mutação Resumo. Se seqüenciaram 36 regiões do genoma de Escherichia coli B escolhidas ao acaso que estão presentes na maioria ou em todos os genomas de E. coli seqüenciados. Se examinaram as relações filogenéticas entre as cepas de E. coli e se buscaram provas da transferência horizontal de genes e se calculou a variação na freqüência de mutação. A árvore filoge nético completo indica que E. coli B e K-12 são as cepas com mais um parentesco estreito, enquanto E. coli ou157:h7 se encontra mais afastada e mais ainda o estão Shigella flexneri 2a e E. coli CFT073, sendo esta última a mais distante de todas. No grupo B, K-12 e O157:H7, várias regiões apóiam topologias alternativas. A transferência horizontal pode explicar essas incongruências filogenéticas. No entanto, também achamos provas de mais uma evolução rápida em lugares sinônimos na linhagem O157:H7. Uma ulterior interpretação destes resultados se confunde por uma associação entre genes que mostram uma evolução mais rápida e os que são transferidos horizontalmente. Mediante o uso de genes do grupo formado por B e K-1, e calculando a velocidade de mutação em um experimento a longo termo com E. coli B e um cálculo de 200 gerações por ano, se estimou que as cepas B e K-12 se separaram há várias centenas de milhares de anos, enquanto Ou157:H7 se separou de seu ancestral comum há entre 1,5 e 2 milhões de anos [Int Microbiol 2005 8(4):271-278]. Palavras chave: cepas de Escherichia coli · evolução experimental · velocidade de evolução · transferência horizontal de genes · evolução molecular |
Introduction
Escherichia coli is one of the most intensively studied living species. While it has long served as a model organism for biochemistry, genetics, and molecular biology, more recently, it has been widely used in experimental studies of evolution. E. coli is a normal part of the microbiota of the lower gastrointestinal tract of mammals, including humans, and usually exists as a harmless commensal. However, there also exist many pathogenic strains of E. coli that can cause a variety of diarrheal and other diseases in humans and animals. These pathogenic strains express virulence factors that are involved in pathogenesis, but which are usually accessory to normal metabolic functions.
Owing to the scientific and clinical interests in E. coli, the genomes of several strains have been completely sequenced, and the sequencing of others is underway. Complete genome sequences are also available for Salmonella strains, which last shared a common ancestor with E. coli more than a hundred million years ago [21]. Differences in ecological strategies and evolutionary histories of the various E. coli strains may have left interesting signatures in their genomes. In the present study, we took advantage of the substantial genomic data available for E. coli to examine some of the mechanisms of molecular evolution that have contributed to the diversity of organisms that microbiologists recognize as E. coli. In particular, we were interested in examining the possible contributions of horizontal gene transfer and differences in mutation rates between strains to genomic differences.
Much of the recent work on E. coli as an experimental system for studying evolution in action has used a derivative of strain B [2,4,14,15,27]. Although a complete genome sequence of E. coli B does not yet exist, sequence data accumulated as part of that research enabled us to examine the phylogenetic relationship between strain B and other E. coli strains with greater precision than was possible previously based on multi-locus enzyme electrophoresis [9] or the genomic distribution of IS elements [28].
The aims of this study were to determine the phylogenetic relationships among various E. coli strains, and to examine several hypotheses that could complicate interpretation of these relationships, including horizontal gene transfer and variation in mutation rates. Thirty-six randomly chosen gene regions of ca. 500 bp each that were previously sequenced in E. coli B were selected and subjected to BLAST searches using the NCBI server. The goal was to find homologous fragments for these genes in seven fully sequenced genomes, including four E. coli strains and three related species: Shigella flexneri, Salmonella typhimurium, and Salmonella enterica. The two Salmonella strains were used in this study primarily for rooting phylogenetic trees. For three of the 36 genes analyzed here (atoA, ycdS, and ycdT), significant homologies were only found within the E. coli genomes; no homologues were found in the Shigella or Salmonella genomes.
Materials and methods
Sequences and alignment. As the starting point for this study, ~500-bp sequences previously obtained for each of 36 randomly chosen and physically dispersed gene regions in E. coli strain B [15] were used. Accessions numbers for these 36 sequences are GenBank:AY625099 to GenBank: AY625134. BLAST searches were then carried out using the NCBI server to find homologous fragments for these genes in seven fully sequenced genomes. The four E. coli strains consisted of one K-12 strain [1], which is a non-pathogenic commensal; two isolates of O157:H7 [16,24], which is enteropathogenic; and one CFT073, which is uropathogenic. The other three genomes were those of Shigella flexneri strain 2a [10], Salmonella typhimurium serovar LT2 [17], and Salmonella enterica serovar Typhi CT18 [23]. As the two O157:H7 isolates are almost identical for all the gene regions included in this study, only the Sakai isolate [16] was used in our analyses.
For each gene, the deduced amino acid sequences were aligned by using the progressive algorithm implemented in CLUSTAL-X [34] and further arranged, as needed, by visual inspection. Alignments for the nucleotide sequences were then obtained from the corresponding protein alignments, thereby ensuring the homology of the sites used in comparisons.
Phylogeny reconstructions. Phylogenetic trees were constructed for each individual gene region, as well as for the concatenated sequence of all of them, by three different methods: neighbor-joining, minimum evolution, and maximum parsimony. The purpose of using three different methods was to evaluate the robustness of the results obtained; in fact, the trees were consistent regardless of the method used. Unless otherwise indicated, Tajima and Nei"s divergence estimator [33] was used for distance-based methods. For all three methods, gap-containing positions were excluded from the analysis. The significance of clusters within the phylogenetic trees was assessed by the bootstrap method with 1000 replicates. Gene trees were consistent regardless of the method used. Phylogenetic trees and molecular evolutionary computations reported in this study were done using MEGA software, version 3.0 [12].
Computation of nucleotide substitution rates. In molecular evolutionary studies, it is generally useful to partition nucleotide substitutions into two classes: (i) nonsynonymous, which replace one amino acid by another, and (ii) synonymous, which leave the same amino acid in the resulting protein. Although most mutations are nonsynonymous, most nonsynonymous mutations are eliminated by natural selection, which causes a numerical predominance of synonymous substitutions. The rates of nonsynonymous and synonymous substitutions were computed following Nei and Gojobori"s modified method and using the correction of Jukes and Cantor for multiple hits [20].
Calculation of codon adaptation index. To test whether an accelerated rate of sequence evolution on a particular branch might reflect changes in codon usage, codon usage tables for each relevant gene in K-12 were calculated using the CUSP utility in the EMBOSS package. These tables were then used to calculate the codon adaptation index CAI [30] for the corresponding genes in strains B and O157:H7 using the CAI utility in EMBOSS.
Results and Discussion
Phylogenetic relationships among E. coli strains. Sequences for each gene region were aligned and the resulting alignments were subjected to independent phylogenetic reconstruction by various methods. In addition, all the gene regions were combined, and an overall phylogenetic tree was constructed from this concatenated sequence. Figure 1 shows the maximum-parsimony (all sites included; branch-and-bound exhaustive search) tree obtained from the concatenated data. The tree has very strong support for all internal branch points based on a bootstrap analysis. This analysis of the entire dataset indicated that strains B and K-12, which are non-pathogenic commensals, are the most closely related pair, followed next by strain O157:H7, which is enterohemorrhagic. Strain CFT073, which is uropathogenic, is the most divergent of the strains within the E. coli clade. Shigella was historically given genus-level status owing largely to its pathology and clinical significance. However, more recent research [9,25] has shown that S. flexneri lies within the E. coli clade, and that it is more closely related to E. coli B, K-12, and O157:H7 than any of these strains are to E. coli CFT073.
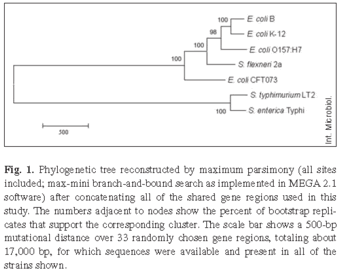
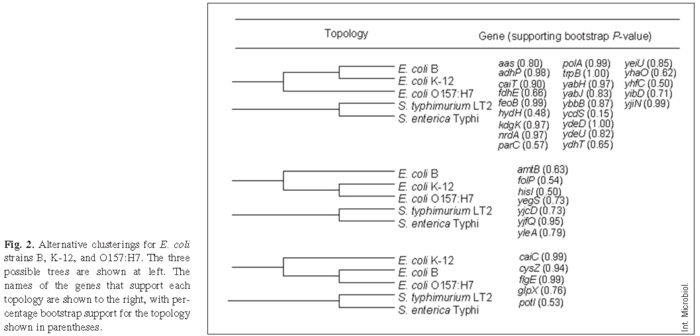
We focused our attention next on the three E. coli strains, B, K-12, and O157:H7, that had diverged more recently from one another and thus formed an internal cluster within the phylogenetic tree. For this analysis, each gene region was examined independently to assess whether it supported the hypothesis that B and K-12 are more closely related to one another than either one is to O157:H7. Alternatively, different genes might support different relationships, which has been reported previously for diverse E. coli strains and which is generally seen as providing evidence for horizontal gene transfer [7,8,18]. Figure 2 shows the three possible topologies relating these strains. The first topology has B and K-12 as the most closely related pair, with O157:H7 more distantly related. Of 35 gene regions that were informative at this level of resolution, 23 supported this clustering (Fig. 2). Of these 23 regions, 16 showed significant bootstrap support at the commonly accepted p ≥ 0.70 level, including nine that fulfilled the very stringent criterion of p ≥ 0.95. The second topology groups K-12 and O157:H7, with B more distantly related. Seven genes supported this topology, including four with bootstrap support at p ≥ 0.70, of which only one fulfilled the very stringent criterion of p ≥ 0.95. The third alternative topology, which pairs B and O157:H7, was supported by five genes, including four significant at p ≥ 0.70 of which two were significant even at the p ≥ 0.95 level. This gene-by-gene analysis therefore tended to support the same topology as that obtained using the entire concatenated sequence (Fig. 1), with strains B and K-12 being more closely related to one another than either is to O157:H7, although several genes provide significant support for alternative topologies. Henceforth, we consider the dominant topology as our null hypothesis, as we seek to explore and better understand these exceptions.
Mechanisms of genomic diversification in E. coli strains. It is well known that horizontal transfer can cause incongruent phylogenies across different genes from the same set of organisms. For example, if species A and B share a recent common ancestor, while species Z is more distantly related; and if a particular genomic segment has recently moved from Z into B; then genes in that segment would support the phylogenetic topology that joins B and Z, whereas the remainder of the genome would support the topology that groups species A and B together. Although this explanation is both logical and plausible, there may be alternative explanations and complications arising from differences in evolutionary rates among genes or across lineages, such as could arise from different selective constraints or mutation rates, respectively. The latter possibility is especially interesting because of some evidence that mutator phenotypes are more common among pathogenic than non-pathogenic E. coli strains [3,13], although a previous sequence-based study found no evidence that O157:H7 had undergone an accelerated rate of molecular evolution [36].
Evidence for horizontal transfer. To examine these issues further, we proceeded as follows. If horizontal transfer is the main explanation for phylogenetic discrepancies among different genes, then the discrepancies should be observed for neutral sites as well as for all sites within those genes. However, if the topology obtained for neutral sites differs markedly from the topology based on all sites within the gene, then alternative explanations should be considered that involve relaxed or otherwise altered selective constraints affecting particular genes in certain lineages. Therefore, minimum evolution trees were recomputed for those eight genes that significantly supported (p ≥ 0.70) the second and third topologies in Fig. 2, but only synonymous sites in the computation of the distance matrix were used. Six of these genes (yegS, yleA, caiC, cysZ, flgE, and glpX) continued to provide significant support (p > 0.70) for their respective alternative phylogenetic topologies, while two (yjcD, yifQ) no longer provided significant support for any topology. Hence, the hypothesis of horizontal transfer involving one or more ancestors of B, K-12, and O157:H7 appears to be well supported by several gene fragments. By contrast, the possibility that certain genes might indicate different topologies owing to altered selective constraints has no compelling support from this analysis, because most genes supporting the non-standard phylogenetic topologies continued to do so when using only synonymous substitutions. However, evidence of gene transfer does not preclude differences in mutation rates between lineages.
Evidence for accelerated evolution in the lineage leading to O157:H7. Two approaches were pursued to test whether the rate of molecular evolution tended to be faster for the lineage leading to O157:H7, presumably owing to this pathogen having spent more of its history as a mutator than did the commensal strains B and K-12. The first approach employed Tajima"s test for unequal rates of molecular evolution [32]. This method is independent of the underlying phylogeny. In our analysis, this test was applied only to third positions in codons in order to minimize any complicating effects of selection. When the test was applied to each individual gene fragment, and using S. flexneri as the outgroup, seven genes (cysZ, flgE, hydH, trpB, ydeD, ydhT, and yeiU) showed significant acceleration (χ2 tests, all p < 0.05) in their rates of neutral evolution along the branch leading to O157:H7. By contrast, only two genes (feoB and yjcD) showed significantly slower neutral evolution along that same branch. When all of the genes were combined and the test was applied to the entire dataset, the results strongly supported an accelerated rate of synonymous substitution on the branch leading to O157:H7 (χ2 = 12.140, 1 df, p < 0.001). These analyses thus support the hypothesis that the pathogenic strain O157:H7 experienced a higher average mutation rate over its history than did two related commensal strains.
It is interesting that the hypothesis of accelerated neutral evolution along the lineage leading to O157:H7 was supported by this analysis but not by a previous analysis by Whittam et al. [36]. One possible explanation for this difference is that our study used a much more closely related outgroup in S. flexneri 2a, whereas the earlier study relied on then-available sequences from the more distantly related Salmonella genome. The inclusion of a much longer period of shared evolutionary history, as would occur with a more distantly related outgroup, might obscure an elevated mutation rate along one branch.
Our second approach to test the hypothesis of accelerated molecular evolution in the branch leading to O157:H7 followed that of Whittam et al. [36]. The synonymous substitution rate, dS, was compared between O157:H7 and S. typhimurium LT2, and between the cluster B + K-12 and S. typhimurium LT2, for each informative gene fragment. Under the null hypothesis of equal rates of evolution along the two branches, a simple linear relationship, with intercept zero and slope one, was expected between the dS values estimated for O157:H7 and B + K-12. Indeed, that simple relationship is what Whittam et al. [36] found using a set of 12 genes for O157:H7 and K-12. Alternatively, if the rate of synonymous substitution was faster along the lineage leading to O157:H7, then the dS values should tend to lie above that isocline. Figure 3 shows the actual relationship between dS values obtained for Salmonella and O157:H7 and those obtained for Salmonella and the cluster containing B and K-12. To test whether there was any significant tendency toward positive deviations from the isocline, we computed for each gene the deviation as the ratio of its dS value on the ordinates axis to its dS value on the abscissas axis, and then subtracted 1. The mean deviation was 0.034 ± 0.012 S.E.M., a value that is significantly greater than zero (t32 = 2.708, 1-tailed p = 0.005) and which therefore further supported the hypothesis of an accelerated rate of neutral evolution on the branch leading to O157:H7. The larger number of genes in our study, compared to the earlier study, provided greater statistical power and might account for the different conclusion. However, as shown below, horizontal gene transfers may further complicate interpretation of these data.

Association of putative horizontal transfers with more rapid evolution. An analysis of variance was carried out to test for possible differences among the alternative phylogenetic topologies in the magnitude of the deviations from the isocline for dS values estimated for O157:H7 versus B + K-12, and a significant effect was obtained (F2,30 = 6.549, p = 0.004). Tukey"s post hoc test further showed that the heterogeneity reflected significant differences between two sets of genes. The first set, which tended to have smaller deviations, included genes supporting the standard topology that groups B and K-12. Notice that the genes in this set are almost evenly distributed around the isocline in Fig. 3, with many points falling above and below it. The second set had a larger mean deviation from the isocline and included those genes supporting the topologies that group either B or K-12 with O157:H7. Notice that 11 of the 12 points in this set lie above the isocline, including four of the five largest deviations in that direction (Fig. 3).
To examine this issue further, we computed the average rates of synonymous and nonsynonymous substitutions for genes classified by the phylogenetic topology they support, using only the branches internal to the three strains. For those genes supporting the topology that groups B and K-12 (n = 20), the average synonymous and nonsynonymous substitution rates were dS = 0.073 ± 0.026 and dN = 0.006 ± 0.002, respectively. For those genes that group K-12 and O157:H7 (n = 7), the corresponding averages were dS = 0.367 ± 0.051 and dN = 0.015 ± 0.006. And for those genes that group B and O157:H7 (n = 5), the average rates were dS = 0.422 ± 0.024 and dN = 0.034 ± 0.007. Tukey"s tests confirmed that both rates were significantly lower for genes that group B and K-12 than for those that support the alternative topologies.
Therefore, there is a consistent association between those genes that support the atypical relationships among B, K-12, and O157:H7 and those that show greater levels of divergence along the branch leading to O157:H7. It is unclear whether this association reflects an important biological process or, alternatively, some artifact that causes statistical confounding between these disparate properties. For example, mutator genotypes (e.g., clones with defects in methyl directed mismatch repair) not only have elevated mutation rates but also are much more prone to incorporating horizontally transferred genes into their chromosomes than strains with functional DNA repair pathways [3,5,26]. However, this association cannot readily explain the pattern seen here, because a particular gene that was transferred from a non-mutator strain into a mutator lineage would be expected to show fewer differences, not more, than genes that remained in the mutator line throughout its hypermutable history. Therefore, the evidence for faster molecular evolution on the branch leading to O157:H7, while intriguing and statistically significant, must be interpreted cautiously until the basis for this association is better understood. In the next two sections, two possible reasons for this association are examined, but a definitive explanation requires further studies.
Acceleration along O157:H7 branch is not caused by changes in codon usage. Codon bias and synonymous substitution rates tend to be negatively correlated because highly expressed genes have more biased codon usage and accumulate synonymous substitutions more slowly owing to selection at the translational level [29]. Therefore, the higher synonymous substitution rate, dS, along the branch leading to O157:H7 could have been a consequence of selection favoring altered codons usage relative to B and K-12. To test this possibility, tables of codon usage by K-12 for each gene, operationally defined the K-12 usage as optimal, were obtained and the codon adaptation index [30], CAI, was then calculated for each corresponding gene in B and O157:H7. More similar codon usage was expected between K-12 and the other strain for genes with higher values of CAI. We already showed that K-12 and B are more closely related than K-12 and O157:H7. Hence, we expected B to have higher CAI values than O157:H7 if the latter has undergone significant changes in codon usage. The average CAI for B is 0.743 ± 0.005 while it is 0.741 ± 0.006 for O157:H7, and the difference is not significant (paired t test: t32 = 0.611, p = 0.546). Therefore, the faster rate of synonymous substitutions along the branch leading to O157:H7 does not reflect any discernible change in codon usage.
Also, there was no significant variation among the genes supporting the different topologies in their GC content at the third positions of codons (ANOVA: F2,30 = 0.044, p = 0.957). The overall average GC content, 60.94 ± 1.06%, is in the range previously reported for E. coli [19]. Differences in GC content sometimes occur for horizontally transferred genes because the donor and recipient strains may have substantially different base composition, if insufficient time has elapsed since the transfer to erode this difference. The only gene that was a conspicuous outlier in terms of its third-position GC content was yibD. In both B and O157:H7, but not in K-12, this gene has an unusually low GC content, although it supports the standard phylogenetic topology that clusters B and K-12 (see Fig. 2).
Acceleration along the O157:H7 branch is not amplified in mismatch repair genes. The rate of sequence evolution appears to have accelerated along the branch leading to O157:H7, which may support the hypothesis that the ancestors of this pathogenic strain spent some of their history as repair-defective mutators [3,13]. If so, the enzymes in the methyl-directed mismatch repair may have evolved especially fast as a consequence of repeated switching between repair-defective and repair-proficient alleles. To test this possibility, we repeated our previous analyses for two mismatch-repair genes, mutS and mutL, whose sequences are available for the relevant strains, including E. coli B [31]. For both genes, the phylogeny inferred from synonymous sites clustered B and K-12 together. If the repair pathway switched repeatedly between mutator and non-mutator states in O157:H7, then the genes encoding that pathway might exhibit an unusually high rate of nonsynonymous substitutions on the O157:H7 branch relative to other genes supporting that phylogeny. The calculated rate of nonsynonymous substitutions was higher for mutL than for 19 of the 23 randomly chosen genes that cluster B with K-12 (Fig. 2); however, mutS has no nonsynonymous substitutions on the O157:H7 branch, ranking it below 11 of the same 23 genes. Thus, the hypothesis that genes encoding the methyl-directed mismatch repair pathway evolved more rapidly along this branch lacks compelling support, although other genes that encode various repair-related functions (e.g., mutH and uvrD) could be involved in such an effect.
Homologous recombination rates, as well as point mutation rates, increase in mutator strains defective for mismatch repair. Hence, it has been hypothesized that restoration of mismatch-repair functions may often occur by gene transfer rather than by back mutation [3,5,26]. However, the fact that both mutS and mutL cluster B with K-12, and the lack of compelling evidence for exceptionally accelerated rates of nonsynonymous substitutions in these genes on the branch leading to O157:H7, provide no more support for this variant hypothesis.
Estimated divergence times between B, K-12, and O157:H7. Based on extensive sequencing of the randomly chosen genes in clones sampled from a long-term evolution experiment, Lenski et al. [15] estimated the mutation rate of repair proficient E. coli B to be about 1.4 × 10–10 mutations per base pair per generation. We therefore also used this value as a proxy for the synonymous substitution rate-under the common assumption that synonymous mutations are neutral and thus substituted at a rate equal to the underlying mutation rate [11,21]-to estimate the approximate times of divergence of E. coli strains B, K-12, and O157:H7. Only those gene regions that supported the B and K-12 cluster with bootstrap values over 0.70 were used in this analysis. These 16 regions yielded 32 synonymous substitutions among 2223 synonymous sites for B and K-12, and 158 or 157 substitutions among the same sites for O157:H7 versus B or K-12, respectively. Combining these values with the above mutation rate, we estimate that B and K-12 diverged -{[log2(1 - 32/2223)]/(1.44 × 10–10)}/2 ≈ 73 million generations ago. Similarly, we estimate that O157:H7 diverged from the ancestor of B and K-12 about 370 million generations ago, or perhaps a bit more recently given the evidence for an elevated mutation rate in the branch leading to O157:H7.
It has been suggested that natural populations of E. coli undergo between 100 and 300 generations per year [21]. Assuming an average of 200 generations per year, the estimates above correspond to about 360 thousand years since the split between the branches leading to B and K-12, and about 1.8 million years since the split that led to O157:H7. Of course, different times would be obtained by using different estimates of the mutation rate. For example, Ochman et al. [21] used comparative data to obtain a mutation rate several fold lower than the value we used, whereas Drake [6] averaged laboratory experiments to obtain a mutation rate several times greater than our value. The absolute times of strain divergence are therefore necessarily rough given the substantial uncertainties about average mutation rates as well as generations per year.
Acknowledgements. This research was supported by a grant from the Spanish MEC-FEDER and a "Salvador de Madariaga" sabbatical grant from the MEC (to SFE); by a grant from the US National Science Foundation (to REL and MAR); and by a grant from the US National Institutes of Health (to TSW). We thank Prof. Dominique Schneider (Grenoble, France) for valuable discussions.
References
1. Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474 [ Links ]
2. Bohannan BJM, Lenski RE (2000) Linking genetic change to community evolution: insights from studies of bacteria and bacteriophage. Ecol Lett 3:362-377 [ Links ]
3. Cebula TA, LeClerc JE (2000) DNA repair and mutators: effects on antigenic variation and virulence of bacterial pathogens. In: Brogden KA (ed) Virulence mechanisms of bacterial pathogens. ASM Press, Washington, DC, pp 143-159 [ Links ]
4. Cooper TF, Rozen DE, Lenski RE (2003) Parallel changes in gene expression after 20,000 generations of evolution in E. coli. Proc Natl Acad Sci USA 100:1072-1077 [ Links ]
5. Denamur E, Lecointre G, Darlu P, Tenaillon O, Acquaviva C, Sayada C, Sunjevaric I, Rothstein R, Elion J, Taddei F, Radman M, Matic I (2000) Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell 103:711-721 [ Links ]
6. Drake JW (1991) A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci USA 88:7160-7164 [ Links ]
7. Dykhuizen DE, Green L (1991) Recombination in Escherichia coli and the definition of biological species. J Bacteriol 173:7257-7268 [ Links ]
8. Guttman DS, Dykhuizen DE (1994) Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science 266:1380-1383 [ Links ]
9. Herzer PJ, Inouye S, Inouye M, Whittam TS (1990) Phylogenetic distribution of branched RNA-linked multicopy single-stranded DNA among natural isolates of Escherichia coli. J Bacteriol 172:6175-6181 [ Links ]
10. Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, Zhang X, Zhang J, Yang G, Wu H, Qu D, Dong J, Sun L, Xue Y, Zhao A, Gao Y, Zhu J, Kan B, Ding K, Chen S, Cheng H, Yao Z, He B, Chen R, Ma D, Qiang B, Wen Y, Hou Y, Yu J (2002) Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucl Acids Res 30:4432-4441 [ Links ]
11. Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, Cambridge, UK [ Links ]
12. Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinf 5:150-163 [ Links ]
13. LeClerc JE, Li B, Payne WL, Cebula TA (1996) High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208-1211 [ Links ]
14. Lenski RE (2004) Phenotypic and genomic evolution during a 20,000-generation experiment with the bacterium Escherichia coli. Plant Breed Rev 24:225-265 [ Links ]
15. Lenski RE, Winkworth CL, Riley MA (2003) Rates of DNA sequence evolution in experimental populations of Escherichia coli during 20,000 generations. J Mol Evol 56:498-508 [ Links ]
16. Makino K, Yokoyama K, Kubota Y, Yutsudo CH, Kimura S, Kurokawa K, Ishii K, Hattori M, Tatsuno I, Abe H, Iida T, Yamamoto K, Onishi M, Hayashi T, Yasunaga T, Honda T, Sasakawa C, Shinagawa H (1999) Complete nucleotide sequence of the prophage VT2-Sakai carrying the verotoxin 2 genes of the enterohemorrhagic Escherichia coli O157:H7 derived from the Sakai outbreak. Genes Genet Syst 74:227-239 [ Links ]
17. McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK (2001) Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852-856 [ Links ]
18. Milkman R (1997) Recombination and population structure in Escherichia coli. Genetics 146:745-750 [ Links ]
19. Muto A, Osawa S (1987) The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci USA 84:166-169 [ Links ]
20. Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418-426 [ Links ]
21. Ochman H, Elwyn S, Moran NA (1999) Calibrating bacterial evolution. Proc Natl Acad Sci USA 96:12638-12643 [ Links ]
22. Ohnishi M, Terajima J, Kurokawa K, Nakayama K, Murata T, Tamura K, Ogura Y, Watanabe H, Hayashi T (2002) Genomic diversity of enterohemorrhagic Escherichia coli O157 revealed by whole genome PCR scanning. Proc Natl Acad Sci USA 99:17043-17048 [ Links ]
23. Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, Sebaihia M, Baker S, Basham D, Brooks K, Chillingworth T, Connerton P, Cronin A, Davis P, Davies RM, Dowd L, White N, Farrar J, Feltwell T, Hamlin N, Haque A, Hien TT, Holroyd S, Jagels K, Krogh A, Larsen TS, Leather S, Moule S, O"Gaora P, Parry C, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG (2001) Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413:848-852 [ Links ]
24. Perna NT, Plunkett G 3rd, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR (2001) Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529-533 [ Links ]
25. Pupo GM, Karaolis DKR, Lan R, Reeves PR (1997) Evolutionary relationships among pathogenic and nonpathogenic Escherichia coli strains inferred from multilocus enzyme electrophoresis and mdh sequence studies. Infect Immun 65:2685-2692 [ Links ]
26. Rayssiguier C, Thaler DS, Radman M (1989) The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342:396-401 [ Links ]
27. Riehle MR, Bennett AF, Lenski RE, Long AD (2003) Evolutionary changes in heat-inducible gene expression in lines of Escherichia coli adapted to high temperature. Physiol Genomics 14:47-58 [ Links ]
28. Schneider D, Duperchy E, Depeyrot J, Coursange E, Lenski RE, Blot M (2002) Genomic comparisons among Escherichia coli strains B, K-12, and O157:H7 using IS elements as molecular markers. BMC Microbiol 2:18. [ Links ]
29. Sharp PM, Li WH (1987) The rate of synonymous substitution in enterobacterial genes is inversely related to codon usage bias. Mol Biol Evol 4:222-230 [ Links ]
30. Sharp PM, Li WH (1987) The codon adaptation index - a measure of directional synonymous codon usage bias, and its potential applications. Nucl Acids Res 15:1281-1295 [ Links ]
31. Shaver AC, Dombrowski PG, Sweeney JY, Treis T, Zappala RM, Sniegowski PD (2002) Fitness evolution and the rise of mutator alleles in experimental Escherichia coli populations. Genetics 162:557-566 [ Links ]
32. Tajima F (1993) Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135:599-607 [ Links ]
33. Tajima F, Nei M (1984) Estimation of evolutionary distance between nucleotide sequences. Mol Biol Evol 1:269-285 [ Links ]
34. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL-X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 24:4876-4882 [ Links ]
35. Welch RA, Burland V, Plunkett G III, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HLT, Donnenberg MS, Blattner FR (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA 99:17020-17024 [ Links ]
36. Whittam TS, Reid SD, Selander RK (1998) Mutators and long-term molecular evolution of pathogenic Escherichia coli O157:H7. Emerging Infectious Diseases 4:1-3 [ Links ]

