










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Ed. impresa)
Print version ISSN 1698-4447
Med. oral patol. oral cir. bucal (Ed.impr.) vol.9 n.1 Jan./Feb. 2004
Tumor fibroso solitario de la región parotidea. Reporte de un caso y revisión de la literatura
María de Lourdes Suárez Roa (1), Luz María Ruíz Godoy Rivera (1), Abelardo Meneses García (1),
Martín Granados-García (2), Adalberto Mosqueda Taylor (3)
(1) División de Patología. Instituto Nacional de Cancerología
(2) Departamento de Cabeza y Cuello. Instituto Nacional de Cancerología
(3) Departamento de Atención a la Salud. Universidad Autónoma Metropolitana Xochimilco. México, D.F.
Correspondencia:
CD María de Lourdes Suárez Roa
División de Patología. Instituto Nacional de Cancerología
Ave. San Fernando 22 Tlalpan México, D.F.
Tel/fax 56 28 04 21
E-mail: lsr3@hotmail.com
Recibido: 14-09-2002 Aceptado: 14-12-2002
| Suárez-Roa ML, Ruíz-Godoy-Rivera LM, Meneses-García A,Granados-García M, Mosqueda-Taylor A. Tumor fibroso solitario de la región parotidea. Reporte de un caso y revisión de la literatura. Med Oral 2004;9:82-8. © Medicina Oral S. L. C.I.F. B 96689336 - ISSN 1137 -2834 |
RESUMEN
El tumor fibroso solitario es una entidad bien establecida, comúnmente localizada en la pleura, aunque recientemente se ha descrito en otras localizaciones, incluyendo la región de cabeza y cuello, en donde se presenta como una lesión bien circunscrita y de crecimiento lento. Debido a su baja frecuencia se puede confundir con otras neoplasias de esta área, por lo que las características microscópicas, ultraestructurales y de inmunohistoquímica ayudan a establecer su diagnóstico. El pronóstico esta relacionado con la localización y el tamaño del tumor. Se presenta un caso en una mujer de 20 años de edad, localizado en la región parotidea con protrusión a cavidad oral, de crecimiento rápido y con un tamaño de 10x8.5x5.5cm, bien circunscrita, y con características histológicas benignas e inmunoreactividad para CD34.
Palabras clave: Tumor fibroso, parótid.
INTRODUCCION
El tumor fibroso solitario (TFS) es una neoplasia poco frecuente, descrita por primera vez en 1931 por Klemperer y Rabin en la pleura, y el cual habitualmente se asocia a superficies serosas (1). En la actualidad se han reportado numerosos casos de TFS en sitios extrapleurales, tales como mediastino, pericardio, retroperitoneo, pulmón e hígado. En la región de cabeza y cuello se ha descrito en sitios no relacionados con cavidades serosas, tales como meninges, fosa infratemporal, órbita, cavidad nasal y senos paranasales, nasofaringe, cavidad oral, glándulas salivales, tiroides, tejido peritiroideo, faringe, espacio parafaringeo y epiglotis (1,2). El TFS se presenta con mayor frecuencia entre la tercera y sexta décadas de la vida, sin tener predilección por género (1,3- 6). La histogenesis del TFS no esta bien definida, ya que aunque inicialmente se pensó que era de origen mesotelial (Stout 1950), estudios ultraestructurales y de inmunohistoquímica sugieren un origen mesenquimal, por mostrar el tumor alta reactividad para este fenotipo y negatividad a antígenos epiteliales (1-3,7). Algunos autores mencionan que el pronóstico de esta entidad se basa en la localización, tamaño y características histológicas del tumor, tales como el aumento de la celularidad, pleomorfismo, alto índice de mitosis, presencia de necrosis y hemorragia, los que se consideran parámetros de malignidad(1,2,5).
En este trabajo se reporta un caso de TFS extrapleural localizado en la región de cabeza y cuello en una mujer joven, en el que se discute su dificultad diagnóstica, patrón histopatológico e inmunohistoquímico, así como su conducta biológica.
CASO CLINICO
Mujer de 20 años de edad, con antecedente de nódulo subcutáneo en la región retroauricular derecha de un año de evolución, asintomática, la que fue extirpada seis meses antes en otra institución sin realizarle estudio histopatológico. La paciente se presenta con aumento de volumen en región parotidea derecha, la cual produce protrusión a cavidad oral, aproximadamente de 6 cm de diámetro, asintomática. La tomografía axial computarizada mostró una masa que se proyecta sobre la región de la fosa pterigomaxilar del lado derecho que comprime y desplaza al espacio parafaringeo de la porción posterior y medial. Se observó bien circunscrita, extendiéndose a paladar duro, fosa infratemporal y obliterando parcialmente el antro maxilar, sin mostrar evidencia de erosión (Figura 1). En la biopsia por aspiración se obtiene material con abundantes células de escaso citoplasma, con núcleos alargados, regulares y cromatina fina, dispuestas en pequeños grupos y sueltas, en un fondo mixoide, lo cual fue considerado compatible con adenoma pleomorfo (Figura 2).

Fig. 1. TAC que demuestra una masa bien delimitada que produce expansión
y desplazamiento de estructuras adyacentes.
CAT shows a well-defined tumoral mass that produces expansion and displaces
adjacent structures.

Fig. 2. BAAF que muestra un grupo de células fusiformes y células dispersas
en una fondo de aspecto mixoide (Papanicolau x100)
Fine needle aspiration biopsy showing a group and isolated fusiform cells
scattered on a myxoid background (Papanicolau x100)
Se realizó parotidectomía superficial derecha y resección de tumor a nivel de la fosa infratemporal. La pieza quirúrgica consistió en dos nódulos, el mayor de forma oval, color blanco, de consistencia firme que midió 10x8.5x5.5 cm. Al corte se observó de aspecto fibroso, sólido, alternando con áreas de tipo mucoide y zonas periféricas hemorrágicas. El nódulo menor midió 1.8 cm de diámetro, y presentó las mismas características que el primero (Figura 3). La glándula parótida resecada no mostró cambios macroscópicos sugestivos de lesión.

Fig. 3. Pieza quirúrgica. Superficie de corte predominantemente sólida, con
áreas periféricas hemorrágicas.
Surgical specimen. Predominantly solid cut surface with peripheral hemorrhagic
areas.
Microscópicamente, el tumor estuvo constituido por células epitelioides ovales, con citoplasma pálido y núcleos alargados hipercromáticos, distribuidas en un estroma levemente mixoide, alternando con áreas hipercelulares, cuyas células eran de aspecto fusiforme, hipercromáticas con núcleos vesiculosos con marcada atípia y nucleolos pequeños ocasionales. Estas zonas alternan con áreas hialinizadas, mostrando un patrón vascular de aspecto hemangiopericitoide (Figura 4). El estudio de inmunohistoquímica reveló intensa positividad para CD34 a nivel citoplasmático (Figura 5), así como para vimentina. La proteína S-100, queratina, desmina y p53 fueron negativas. Lo anterior confirmo el diagnóstico de TFS. La paciente recibió radioterapia postoperatoria de 65 Gy a la zona afectada, y un año después se encuentra sin actividad tumoral.

Fig. 4. Áreas hipercelulares de células fusiformes en patrón desordenado, zonas
de aspecto hemangiopericitoide y fondo levemente mixoide (H.E. x100).
Hypercellular areas composed of spindle cells and hemangiopericytoid-like
areas over a slightly myxoid background (H.E. x100).
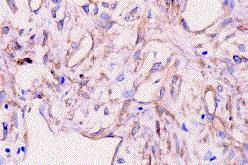
Fig. 5. Células tumorales y endotelio vascular altamente positivos para
CD 34 (x400).
Highly positive CD34 tumoral cells and vascular endothelium (x400).
DISCUSION
El TFS es una neoplasia rara en la región de cabeza y cuello, de crecimiento lento, bien circunscrita y con bajo índice de recurrencia. La edad de presentación esta entre los 30 y 70 años, sin predilección por género. Los síntomas básicamente están relacionados con la localización y el tamaño de la lesión, y habitualmente se resuelven con su extirpación (1,8,9). En cabeza y cuello esta lesión reviste gran interés, ya que al no comprometer alguna estructura importante en su etapa inicial puede pasar desapercibida, alcanzado así un tamaño considerable o invasión a estructuras vecinas, lo que hace difícil en muchas ocasiones su extirpación completa. En la tabla 1 se presentan las características clínicas sobresalientes de los casos previamente reportados de TFS en cavidad oral y región maxilofacial

Tabla 1. Características generales de los casos reportados de TFS de la región
oral y maxilofacial
Basado en referencias (1, 3-5, 7, 9, 12-15, 18)
* NR. No referido
El caso aquí presentado alcanzó un tamaño de 10 cm, provocando dolor por compresión. Los casos reportados a nivel de cavidad oral han sido de menor tamaño, lo que podría explicarse por las dimensiones propias de la cavidad oral y la sintomatología temprana que se presenta por interferir con las funciones de esa región, lo que difiere con otras regiones de cabeza y cuello, tal como cavidad nasal, donde se han reportado casos de hasta 7cm, en los que el principal síntoma es obstrucción nasal y rinorrea (9) . En la región de cabeza y cuello estos volúmenes son habituales en neoplasias malignas y en algunas lesiones benignas con largo tiempo de evolución. La invasión a estructuras adyacentes es importante porque esta situación puede provocar la extirpación incompleta del tumor. En nuestro caso, al ser bien circunscrito su extirpación fue completa en ambos nódulos.
Uno de los aspectos más importantes lo constituye el diagnóstico diferencial de esta lesión, tanto a nivel clínico como histopatológico. En el presente caso la primera consideración diagnóstica clínica fue de adenoma pleomorfo versus un tumor mixto maligno, por encontrarse el tumor en la región parotídea así como por el tiempo referido de evolución que fue de seis meses y el antecedente de extirpación previa de una lesión en esta zona. Estas posibilidades diagnósticas fueron apoyadas por la BAAD, la que se consideró compatible con adenoma pleomorfo. Con respecto al TFS, son pocos son los reportes por BAAD descritos en la literatura, y la mayoría de estos han sido descritos en pleura. En un reporte de 6 casos presentado por Clayton et al (10), incluyendo uno extrapleural y dos malignos, estos autores describen las siguiente características citológicas: fondo poco hemorrágico y escasas células inflamatorias, células poligonales y fusiformes predominantemente en un patrón en células individuales con núcleos ovales o redondos, citoplasma fino, cromatina finamente granular y uniforme. Esta descripción es muy parecida a lo que se observa en el adenoma pleomorfo por BAAD (11). En estos casos resulta muy difícil establecer el diagnóstico de TFS por BAAD, ya que la variedad citológica del tumor y su presentación en parótida, frecuentemente llevan al diagnóstico de adenoma pleomorfo u otras entidades con características citológicas similares en otras localizaciones (4,5,10,12). Clayton et al (10), al igual que otros autores, enfatizan la importancia del corte histopatológico para el diagnóstico definitivo.
Macroscópicamente el TFS se describe como una neoformación bien circunscrita, no encapsulada, de superficie externa lisa de color blanco, con áreas translucidas y consistencia firme, tal como se observó en este caso (1-3,12,13), pero también con características macroscópicas muy similares al adenoma pleomorfo, aunque en la mayoría de las ocasiones esta entidad esta con relación a la glándula y se pueden observar varios nódulos. En el presente caso ambos nódulos se encontraban inmersos en los tejidos blandos sin relación alguna con el tejido glandular.
En 1997 Chan (2) propuso como esenciales los siguientes criterios diagnósticos para el tumor fibroso solitario: que sea circunscrito, la presencia de áreas hipercelulares alternando con zonas escleróticas hipocelulares, aspecto inocente, aspecto fusiforme corto, células ovoides con escaso citoplasma pobremente definido, pocas mitosis (<4/10 campos de alto poder), células alargadas dispuestas en forma desordenada, fasicular o estoriforme y entrelazadas íntimamente con fibras de colágeno, lo que coincide con lo descrito por otros autores (1,2,11-16). Como hallazgos secundarios, se pueden encontrar calcificaciones, cuerpos de psammoma, así como áreas focales con cambios mixoides ( 2,17).
Los estudios de inmunohistoquímica ayudan a establecer el diagnóstico. Las células ovoidales de aspecto epitelioide no expresan proteína S100, desmina, citoqueratina o EMA. El anticuerpo CD34 es intensamente positivo en la mayoría de los estudios reportados, y para Chan (2) es un criterio necesario. Aunque para algunos autores no es considerado así, este hallazgo es un apoyo fundamental para el diagnostico de TFS extrapleural (3,7,12,15,18).
La presencia de áreas sarcomatosas (hipercelularidad, atípia nuclear, pleomorfismo y mas de 4 mitosis/10 campos de alta resolución) dentro del TFS típico, o el desarrollo de un sarcoma en el mismo sitio donde se presento un TFS, son dos condiciones satisfactorias como criterios de malignidad, aunque algunos autores no están de acuerdo y básicamente lo consideran cuando hay recurrencia y metástasis a distancia (2,7,16). El reporte de malignidad en pleura es del 23%, pero esta característica en los tumores extrapleurales es rara (9-11). Nuestro caso presentó características clínicas e histológicas malignas y benignas como atipia nuclear y pleomorfismo, crecimiento relativamente rápido y gran tamaño, y a su vez estuvo bien delimitado y careció de mitosis.
En un trabajo realizado por Yokoi et al (16) en 10 casos, estos autores encontraron que el TFS maligno puede ocurrir de novo o por transformación a partir de una lesión benigna o de bajo grado y puede estar asociado con mutación en p53 y que a pesar que la proteína CD34 es el marcador usual para este tumor, no logra mantenerse esta expresión en los tumores de alto grado, ya que ellos encontraron positividad de esta proteína en TFS maligno de bajo grado y grado intermedio y negatividad para los de alto grado. Hanau y Miettinen reportaron en un estudio de 29 TFS, tres casos malignos sin especificar grado, positivos para CD34. (7) Nuestro caso presentó alta positividad para CD34 y negatividad para p53, por lo que, basándonos en lo reportado por la literatura, nosotros consideramos que puede tratarse de una neoplasia maligna de bajo grado (7,16).
El diagnostico diferencial del TFS en cabeza y cuello debe hacerse con fibrosarcoma, histiocitoma fibroso maligno o benigno, metástasis de mesotelioma maligno, hemangiopericitoma y tumores de la vaina nerviosa benignos y malignos (2,8,11,15,18). Sin embargo, en la mayoría de los casos el patrón morfológico y los hallazgos de inmunohistoquímica permiten descartar estas neoplasias. Así, el histiocitoma fibroso presenta un patrón estoriforme y marcado pleomorfismo celular con positividad para S100, que también es positivo para los tumores benignos y malignos de la vaina nerviosa en los cuales se pueden presentar áreas mixoides (2,17,18)
El tratamiento de elección para el TFS es la resección quirúrgica completa. Algunos autores han reportado resultados favorables con quimioterapia y radioterapia después de cirugía en caso de resecciones incompletas (9,15). El tumor fibroso solitario puede recurrir después de muchos años, por lo que la resección completa es el factor pronóstico más importante (8,11). El comportamiento biológico del TFS es impredecible, ya que el 23% de los TFS en pleura han manifestado agresividad (1,15, 18). Algunos autores consideran que la invasión, recurrencia y las metástasis a distancia pueden estar relacionadas con el tamaño del tumor, la hipercelularidad, el incremento de mitosis, pleomorfismo e historia de hemorragia (8).
CONCLUSION
El TFS en cavidad oral y región maxilofacial a pesar ser poco frecuente, es una entidad a considerar en el diagnóstico diferencial de las lesiones que pueden presentar características clínicas e histológicas similares, de las que se debe diferenciar mediante estudios de inmunohistoquímica. El pronostico del TFS en cabeza y cuello es reservado por los pocos casos reportados hasta la fecha, pero se debe basar en la localización y tamaño de la neoplasia. El rápido crecimiento, la recurrencia y las metástasis, son factores determinantes para considerarlo maligno.
BIBLIOGRAFÍA/REFERENCES
1. Rayappa CS, McArthur PD, Gandopadhyay KA. Solitary fibrous tumor of the infratemporal fossa. J Laryngol Otol 1996;110: 594-7 [ Links ]
2. Chan JKC. Commentary. Solitary fibrous tumour-everywhere, and a diagnosis in vogue. Histopathology 1997;31:568-76 [ Links ]
3. Stringfellow HF, Khan FR, Sissons MC, Path MR. Solitary fibrous tumor arising in the nasal cavity: report of a case. J Laryngol Otol 1996;110:468-70. [ Links ]
4. Zukerberg LR, Rosenberg AE, Randolph G, Pilch BZ, Goodman ML. Solitary fibrous tumor of the nasal cavity and paranasal sinuses. Am J Surg Pathol 1991;15:126-30. [ Links ]
5. Günhan Ö, Yildiz RF. Celasun B, Önder T. Finci R. Solitary fibrous tumour arising from sublingual gland: report of a case. J Laryngol Otol 1994;108:998-1000. [ Links ]
6. Al-Sinawi A, Johns MA. Parapharyngeal solitary fibrous tumour: an incidental finding at ENT examination. J Laryngol Otol 1994;108:344-7. [ Links ]
7. Hanau CA, Miettinen M. Solitary Fibrous Tumor: Histological and inmmunohistochemical spectrum of benign and malignant variants presenting at different sites. Hum Pathol 1995;26:440-9. [ Links ]
8. Goodland JR, Fletcher CDM. Solitary fibrous tumour arising at unusual sites: analysis of a series. Histopathology 1991;19:515-52. [ Links ]
9. Witkin GB, Rosai J. Solitary fibrous tumor of the upper respiratory tract. Am J Surg Pathol 1991;15:842-8. [ Links ]
10. Clayton AC, Salomao DR, Keeney GL, Nascimiento AG. Solitary fibrous tumor: a study of cytologic feactures of six cases diagnosed by fine-needle aspiration. Diagn Cytopathol 2001;25:172-6 [ Links ]
11. Rojas TME, Moreno LH, Rodríguez VA. Biopsia por aspiración con aguja delgada de glándulas salivales. En: Angeles AA. Biopsia por aspiración con aguja delgada. México: Angeles Editores;1994. p 76-7 [ Links ]
12. Talacko A, Aldred MJ, Sheldon WR, Hing NR. Solitary fibrous tumor of the oral cavity: report of two cases. Pathology 2001;33:315-8. [ Links ]
13. Alawi F, Stratton D, Freedman PD. Solitary fibrous tumor of oral soft tissues. A clinicopathologic and immunohistochemical study of 16 cases Am J Surg Pathol 2001;25:900-10. [ Links ]
14. Kurihara K, Mizuseki K, Sonobe J, Yanagihara J. Solitary fibrous tumor of the oral cavity. Report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;87:223-6. [ Links ]
15. Perez OB, Koutlas IG, Strich E, Gilbert RW, Jordan RC. Solitary fibrous tumor of the oral Cavity. An uncommon location for a ubiquitous neoplasm. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;87:589-93. [ Links ]
16. Yokoi T, Tsuzuki T, Yatabe Y.Suzuki M, Kurumaya H, Koshikawa T,et al. Solitary fibrous tumour: significance of p53 and CD34 inmmunoreactivity in its malignant trasformation. Histopathology 1998;32:423-32. [ Links ]
17. Iwai S, Nakazawa M, Yoshikawa F, Amekawa S, Sakuda M. Solitary fibrous tumor of the buccal mucosa. Report of a case with immunohistochemical studies. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;88:461-5. [ Links ]
18. Vargas PA, Alves FA, Lopes MA, Siqueira SAC, Menezes LFC, Aldred VL, et al. Solitary fibrous tumour of the mouth: report of two cases involving the tongue and cheek. Oral Dis 2002;8:111-5. [ Links ]

