










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Ed. impresa)
Print version ISSN 1698-4447
Med. oral patol. oral cir. bucal (Ed.impr.) vol.9 n.2 Mar./Apr. 2004
Oral manifestations of chronic disseminated histiocytosis.
A report of 10 cases
MÍNGUEZ I, MÍNGUEZ JM, BONET J, PEÑARROCHA M, SANCHIS JM. ORAL MANIFESTATIONS OF CHRONIC DISSEMINATED HISTIOCYTOSIS. A REPORT OF 10 CASES. MED ORAL 2004;9:149-54.
SUMMARY
Chronic disseminated histiocytosis is a systemic disorder resulting from tumor proliferation of Langerhans-type histiocytic cells. The etiology and pathogenesis are not fully clear, though the clinical manifestations are the result of the accumulation and infiltration of these types of cells in organs and tissues. The present study reports 10 patients (6 boys and 4 girls) with chronic disseminated histiocytosis. The patient age at onset of the disease varied from 4 months to 3.2 years (mean 1.7 years). All patients had oral lesions, and in 5 cases these were the first manifestation of the disease. The most frequent alterations were gingival bleeding (7 cases), aphthae measuring over 1 cm in diameter (6 cases), maxillary osteolytic lesions (6 cases), tooth loss due to expulsive folliculitis (5 cases), oral candidiasis (4 cases), orofacial swelling (3 cases), aphthae measuring under 1 cm in diameter (3 cases), and nonspecific oral pain (2 cases). All the oral lesions disappeared with the treatments prescribed, though some patients developed new outbreaks and exacerbations of the disease.
Key words: Hand-Schüller-Christian syndrome, chronic disseminated histiocytosis, oral.
INTRODUCTION
Chronic disseminated histiocytosis (CDH), or Hand-Schüller-Christian syndrome, is classified along with eosinophilic granuloma and Letterer-Siwe disease as a form of histiocytosis X or Langerhans-cell histiocytosis. The disorder involves the proliferation of Langerhans-type histiocytic cells, with different clinical features ranging from localized to generalized presentations (either acute or chronic), with the involvement of bone, organs and the skin (1,2).
The etiology and pathogenesis of CDH and of the other forms of Langerhans-cell histiocytosis remains unclear, though according to Steward et al. (3), the cells that proliferate in these lesions share immunohistochemical and ultrastructural characteristics with Langerhans cells. The associated systemic alterations result from the accumulation of Langerhans cell infiltrates which produce different clinical manifestations depending on their location.
CDH has classically been defined as a triad comprising diabetes insipidus, exophthalmos and bone alterations. However, the disseminated form of the disease can also affect other structures of the reticuloendothelial system (RES), such as the lymph nodes, liver or lungs. The condition tends to manifest in the first decade of life, at around three years of age, and no particular sex predilection has been identified (2,4,5).
The present study reports a series of 10 children with CDH, with special emphasis on the oral alterations of the disease, which may constitute the first manifestation of the syndrome.
CLINICAL CASES
The present study comprises 10 children (4 girls and 6 boys) with a mean age of 19.1 months at the time of diagnosis (range 4-36 months). All were initially diagnosed with chronic disseminated histiocytosis (CDH), though in one patient (case 7) the disease adopted an acute course 7 years after diagnosis, leading to death at age 11 years due to inoperable cranial infiltration.
In five patients (50%) the oral lesions were the first manifestation of the disease, and fundamentally consisted of tooth mobility and loss, gingival bleeding, aphthae and ulcerations. X-ray findings were recorded in 6 patients, in the form of radiotransparent zones with osteolytic areas resulting from infiltration and destruction of the mandibular and maxillary structures by histiocytic tumor infiltrates (Fig. 1A and B). In the course of the disease other oral manifestations were also diagnosed, including candidiasis, nonspecific pain, orofacial swelling and maxillary osteolytic lesions attributable to histiocyte invasion (Table 1).

Table 1. Clinical characteristics of the ten patients with chronic disseminated histiocytosis. Treat. = treatment provided,
S = surgery, CT = chemotherapy, RT = radiotherapy, SC = systemic corticoids, IC = intralesional corticoids, M = male, F = female
The most frequent systemic manifestations were skin lesions, alterations of the long bones (Fig. 2) and skull, diabetes insipidus, exophthalmos, pulmonary involvement, otitis, ferropenic anemia and fever (Table 2).
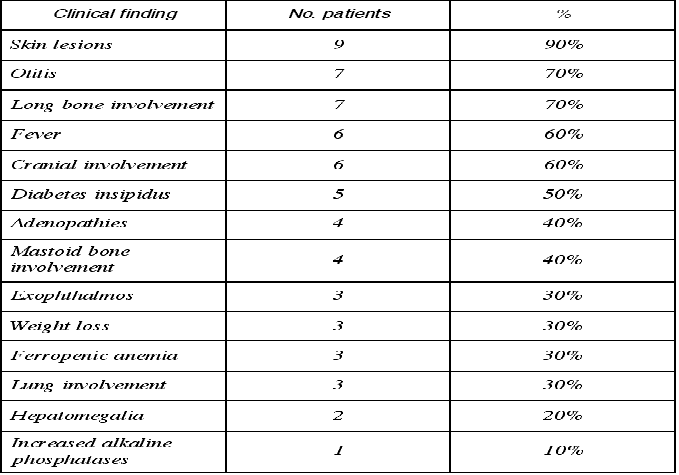
Table 2. Extraoral clinical manifestations of chronic disseminated histiocytosis.
All patients received chemotherapy in the form of vinblastine (6 mg/m2/week in 6 cycles) followed by maintenance therapy with mercaptopurine (50 mg/m2/day) and methotrexate (20 mg/m2/week)(both via the oral route). In 8 patients this regimen was associated to corticoids (100 mg/m2/day for 3 days, followed by gradual dose tapering to 7 mg/m2/day). Four patients were subjected to surgery for the removal of maxillary histiocytic infiltrates, while two patients (cases 7 and 10) underwent radiotherapy. One patient (case 9) received intralesional corticotherapy in the form of methylprednisone (50 mg, 8 doses on alternate days).
After an average follow-up of 6 years (range 1-11 years), two cases were seen to have remitted completely, 6 presented periodic relapses followed by stabilization, one patient died due to inoperable cranial involvement, and one patient was lost to follow-up after one year.
DISCUSSION
Chronic disseminated histiocytosis (CDH) is an infrequent disorder of childhood. According to Bagán et al. (2), the approximate incidence is one case in every 3,300,000 individuals. The disease usually manifests in the first decade of life, between ages 3 and 10 years, with no particular sex predilection (4). The present study comprised 6 boys and 4 girls, with an average age of 1.7 years at the time of diagnosis.
CDH has classically been defined as a triad comprising diabetes insipidus, exophthalmos and bone alterations, though only 25% of patients present the full picture (2,6), and according to García-Pola et al. (7) the complete triad is actually found in less than 10% of cases. In only one of our patients (10%) was the full triad identified, while two patients presented two of the alterations (3 with bone lesions and diabetes insipidus, and 2 with bone lesions and exophthalmos).
Regarding the systemic manifestations of CDH, bone and skin alterations are often observed, with less frequent visceral involvement (8). The most commonly affected bones are the skull and facial bones, the femur, ribs, vertebras and humerus. In general, any bone in the body can be affected, though the bones of the hands and feet are only rarely involved (9). Ninety percent of our patients had bone lesions. In turn, 50% showed simultaneous infiltration of the long bones and skull, while two patients only presented long bone lesions and two children had only cranial infiltrates. The skin was affected in 42% of the patients examined by Sigala et al. (4), and in 12% of the 114 subjects described by Hartman (5). Ninety percent of our series presented skin lesions - the scalp being the most frequently affected location (40%). Regarding other typical manifestations of the syndrome, we identified otitis media in 70% of cases. In comparison, otitis media has been reported in the literature in between 11% (5) and 38% (4) of cases. Twenty percent of our patients presented hepatomegalia, compared with 8% according to Hartman (5) and 42% in the series by Sigala et al. (2,9).
According to Blaschke (10), oral lesions often constitute the first manifestation of CDH. Shaw et al. (11) described the first symptoms referred by their patients: facial swelling, gingival necrosis and loss of alveolar bone reminiscent of juvenile periodontitis or localized marginal periodontitis. Sigala et al. (4), in a series of 50 cases, found oral manifestations of the disease in 36% of patients, while in 16% of cases the first manifestation affected the orofacial region. In one of our patients the syndrome was diagnosed after detecting whitish gingival plaques, bleeding of the gums and tooth mobility. In four cases CDH simultaneously presented oral and systemic alterations, while in 5 cases and following the systemic alterations, oral symptoms developed in the course of the disease. These observations suggest that the oral manifestations of CDH are important, and sometimes generate diagnostic problems since they may simulate periodontitis (11), which exhibits very similar symptoms: local pain, gingival swelling and deep bursae accompanied by alveolar bone loss. Such manifestations in very young patients should cause us to suspect the existence of an underlying or background disorder such as histiocytosis (11,12).
The most widely used treatment options comprise chemotherapy, radiotherapy, surgery and corticotherapy (both systemic and intralesional). No consensus exists over the best treatment combination, and the choice of management approach is largely dependent upon the location and spread of the disease (2). Different therapeutic approaches are normally required, according to the biological behavioral changes which tend to characterize CDH (2,5,8-10). In those cases where a localized lesion is found, surgical removal is indicated (2,7). Only four of our patients (cases 6, 7, 9 and 10) required surgery for the elimination of oral lesions - the combination of chemotherapy and systemic corticoids being the most often prescribed treatment approach, with dose variations according to the outbreaks of the disease. The oral lesions disappeared in all cases.
As regards patient evolution, the disease is characterized by frequent relapses in relation to the type of treatment provided. In this context, the association of surgery plus irradiation or chemotherapy involves fewer relapses than irradiation or surgery alone (5). Curiously, in our series, the patient subjected to surgery with chemo- and radiotherapy was the only subject who suffered an acute outbreak and died as a result of cranial spread of the disease. However, the combination of chemotherapy and corticoids proved effective in the rest of our series.
These patients must be subjected to periodic revisions, for although the oral lesions heal with treatment, the course of the disease is unpredictable and involves outbreaks - the prognosis being particularly unfavorable when spread is associated with dysfunction at liver, lung or hematopoietic level (3-5,7).
REFERENCES
1. Gnanaskhar JD, Ahmad MS, Reddy RR. Histiocitosis de células de Langerhans multifocal de la mandíbula. Presentación de un caso. Quintessence 1992;8:482-5. [ Links ]
2. Bagán JV, García-Pola MJ. Histiocitosis X en la cavidad oral. En: Bagan JV, Ceballos A, Bermejo A, Aguirre JM, Peñarrocha M. Medicina Oral. Barcelona: Masson; 1995. p. 470-1. [ Links ]
3. Stewart JC, Regezi JA, Lloyd RV, McClatchey KD. Immunohistochemical study of idiophatic histyocitosis of the mandible and maxila. Oral Surg Oral Med Oral Pathol 1986;61:48-53. [ Links ]
4. Sigala JL, Silverman S, Brody HA, Kushner JH. Dental involvement in histiocytosis. Oral Surg Oral Med Oral Pathol 1972;1:42-8. [ Links ]
5. Hartman K. Histiocytosis X: a review of 114 cases with oral involvement. Oral Surg Oral Med Oral Pathol 1980;1:38-54. [ Links ]
6. Guillén C. Histiocitosis. En: Guillén C, Botella R, Sanmartín O, eds. Manual Janssen de enfermedades de la piel. Barcelona: Masson; 1996. p. 142-6. [ Links ]
7. García-Pola MJ. Enfermedad de las células de Langerhans. En: Echeverría JJ, Cuenca E, eds. El Manual de Odontología. Barcelona: Masson 1995. p. 153-4. [ Links ]
8. Siew Tin O, Chin Boon L. Chronic disseminated histiocytosis X: a case report. J Clin Pediatric Dentistry 1992;1:33-5. [ Links ]
9. Pacino GA, Serrat A, Redondo LM, Verrier A. Histiocitosis de Células de Langerhans: aspectos clínico-diagnósticos y conceptos actuales. Medicina Oral 1999;5:607-18. [ Links ]
10. Blaschke D. Enfermedades sistémicas que se manifiestan en los maxilares. En: Goaz-White. Radiología oral, principios e interpretación. Madrid: 1995. [ Links ]
11. Shaw L and Glenwright HD. Histiocytosis X: an oral diagnostic problem. J Clin Periodontol 1988;15:312-5. [ Links ]
12. Artzi Z, Gorsky M, Raviv M. Periodontal manifestations of adult onset of Histiocytosis X. J Periodontol 1989;1:57-66. [ Links ]

