










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Ed. impresa)
Print version ISSN 1698-4447
Med. oral patol. oral cir. bucal (Ed.impr.) vol.9 n.3 May./Jul. 2004
Neurofibroma plexiforme en mucosa yugal:
Presentación de un caso clínico
Guillermo Gómez Oliveira(1), Javier Fernández-Alba Luengo(2), Roberto Martín Sastre(2),
Beatriz Patiño Seijas(1), José Luis López-Cedrún Cembranos(3)
(1) Médico Residente
(2) Médico Adjunto
(3) Jefe de Servicio. Servicio de Cirugía Maxilofacial del CHU Juan Canalejo (La Coruña). España
Correspondencia:
Dr. Guillermo Gómez Oliveira
Xubias de Arriba, 84 15006 La Coruña
Teléfono: 981 17 80 00 Extensión 20113
E-mail: guis@canalejo.org
Recibido: 29-06-2003 Aceptado: 07-12-2003
| Gómez-Oliveira G, Fernández-Alba-Luengo J, Martín-Sastre R, Patiño-Seijas B, López-Cedrún-Cembranos JL. Neurofibroma plexiforme en mucosa yugal: Presentación de un caso clínico. Med Oral 2004;9:263-7. © Medicina Oral S. L. C.I.F. B 96689336 - ISSN 1137 - 2834 |
RESUMEN
Presentamos un caso clínico de neurofibroma plexiforme localizado en región geniana, a nivel submucoso. Su interés radica en que, a pesar de ser el tumor de origen neurógeno más frecuente, es una entidad poco habitual y que rara vez se localiza a nivel intraoral. Por otra parte, la variedad plexiforme es todavía menos frecuente. Desde el punto de vista clínico, se manifiestan como lesiones anodinas, con escasa sintomatología, que cuando aparece es derivada de la compresión nerviosa. En nuestro caso el tumor era asintomático salvo por el tamaño. Radiológicamente no existe una imagen definitiva. Tiene relación con determinados síndromes poliglandulares y facomatosis. El tratamiento es básicamente quirúrgico aunque existen dudas de la idoneidad del mismo y se están buscando nuevas vías de tratamiento. Aprovechando la descripción del caso se realiza una revisión bibliográfica incidiendo en la epidemiología, comportamiento clínico, métodos diagnósticos, así como en el tratamiento de este tipo de tumores benignos.
Palabras clave: Neurofibroma plexiforme, neurofibromatosis, tumor benigno, facomatosis, Von Recklinghausen
INTRODUCCIÓN
neurofibromas son proliferaciones benignas que derivan de los nervios periféricos (1-7) y representan uno de los tumores de origen neurógeno más frecuentes (7,8). Su origen parece estar en relación con las células de Schwann y los fibroblastos perineurales (3,6,7,9).
Se han descrito presentaciones múltiples (1-4,6-8,10) o solitarias. Los primeros aparecen muy frecuentemente asociados a neurofibromatosis,(7,8,10,11) y son, por tanto, muy sugestivos de esta enfermedad. Los solitarios son más raros y pueden presentarse asociados ó no a este síndrome (2,3,8).
La cavidad oral no es asiento habitual de neurofibromas solitarios, pero de darse aquí, la localización más frecuente suele ser la lengua, aunque también pueden verse en paladar, mucosa yugal, suelo de boca e incluso intraóseo a nivel mandibular (2-4,7).
El aspecto clínico es el de un nódulo pediculado ó sesil, de crecimiento lento, y que puede afectar tanto a piel como a mucosas (2,3,6,7). Esto obliga, en ocasiones, a hacer un diagnóstico diferencial con lesiones de aspecto similar como papilomas, hiperplasias fibrosas y schwannomas (2,12).
La sintomatología que producen deriva generalmente de la compresión nerviosa (4), y se manifiesta como parestesias o dolor.
Histológicamente se diferencian dos variedades según su aspecto microscópico: mixoide y plexiforme (2,3). El primero es el más frecuente, mientras que el segundo no es tan común (9) y puede aparecer asociado a síndromes poliglandulares (MEN III) (13) y a facomatosis del tipo de la enfermedad de Von Recklinghausen.
El tratamiento de elección es la exéresis quirúrgica, aunque hay autores como Gupta y cols, que están comenzando a emplear una nueva terapia con talidomida en pacientes portadores de neurofibromas plexiformes sintomáticos, ante las dudas que plantea el tratamiento quirúrgico (14).
CASO CLÍNICO
Mujer de 67 años de edad portadora de prótesis de rodilla y con antecedentes de colecistectomía y herniorrafia umbilical, que acude a consulta por presentar tumoración en región geniana izquierda de unos 2 años de evolución, que ha ido disminuyendo de tamaño muy lentamente. La única sintomatología referida es la derivada del tamaño del tumor.
En la exploración se aprecian dos tumoraciones, aparentemente independientes entre sí y de la parótida, de unos 2 ó 3 cm, móviles, bien definidas y de consistencia elástica.
Se realiza PAAF preoperatoria que muestra un frotis con cuadro citológico sugestivo de tumor mixto de glándula salival. Dado que en la exploración la tumoración se localiza, en principio, independiente de la parótida y en una localización poco frecuente para un adenoma pleomorfo, se decide realizar RNM. Esta evidencia una lesión ocupante de espacio en partes blandas de la mejilla izquierda, de señal homogénea, hipointensa en T1 y marcadamente hiperintensa en T2 (Fig. 1). Esta imagen es compatible con linfangioma, que en su parte craneal se extiende hasta la altura del límite superior de la órbita y desciende en contacto con el borde externo del masetero hasta la altura de la cavidad bucal donde lo bordea anteriormente y se introduce medialmente contactando con el maxilar superior y la pared externa del seno maxilar.

Fig. 1. MRI shows an intense lesion.
La Resonancia nuclear magnética evidencia
una lesión claramente intensa
Tras realizar preoperatorio, se practica la intervención mediante un abordaje intraoral. Al incidir la mucosa se observa una formación que se hernia, y que resulta fácil de separar de los tejidos circundantes mediante disección roma (Fig. 2). El resultado final nos muestra una tumoración de 2 x 7 cm, de aspecto quístico y forma tubular arrosariada (Fig. 3).

Fig. 2. Intraoperative view of the tumor through
the incision.
Vista intraoperatoria de la tumoración herniada
a través de la incisión.
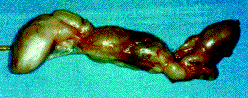
Fig. 3. Operative specimen.
Pieza quirúrgica.
El estudio anatomopatológico de la pieza extirpada muestra macroscópicamente una formación elongada, cilíndrica, de unos 11 x 1,7 cm cubierta por una cápsula con vasos prominentes en superficie y estructuras cilíndricas adheridas a la formación principal. Al corte presenta áreas de aspecto fibroso denso, hialinizado, alternando con zonas de aspecto más laxo. El estudio microscópico reveló una proliferación de células con citoplasma eosinófilo, de bordes mal definidos, con núcleos fusiformes con sus extremos elongados, que se disponen en haces adquiriendo a veces un aspecto serpenteante de las células. No se observan figuras de mitosis. La tumoración presenta zonas más densas que alternan con otras mucho más laxas, contiene vasos de paredes a veces hialinizadas, en general finas y algunas dilatadas quísticamente. La tumoración aparece bien delimitada y en la cápsula se observan fascículos nerviosos, algunos con comienzo de proliferación. El diagnóstico anatomopatológico definitivo fue el de neurofibroma plexiforme.
En nuestro caso no encontramos ningún hallazgo clínico ni antecedente personal ó familiar sugestivo de neurofibromatosis tipo I ó de síndrome poliglandular MEN III (MEN IIb) y parece confirmar que las lesiones aisladas son independientes de estos síndromes.
En la actualidad, dos años después de la intervención, la paciente no ha presentado ningún signo de recidiva tumoral en las sucesivas revisiones.
DISCUSIÓN
El neurofibroma plexiforme es un tumor benigno de origen mesenquimatoso, que se desarrolla a partir de las células de Schwann y los fibroblastos perineurales de los nervios periféricos (6). Es una entidad poco frecuente, aunque constituye la neoplasia mas frecuente de los nervios periféricos (7). Es un tumor que presenta asociación con la enfermedad de von Recklinghausen y con el síndrome poliglandular MEN III, por lo que, ante su presencia, habría que realizar un despistaje de estas dos entidades (4) (Tablas I y II).

Table I. Diagnostic criteria for Type I neurofibromatosis (From Neurofibroma de la mucosa alveolar.
A propósito de un caso. 1998) (15)
Tabla I. Criterios diagnósticos de neurofibromatosis Tipo I (Tomado de Neurofibroma de la mucosa alveolar.
A propósito de un caso. 1998) (15)

Table II. Clinical manifestations for MEN III (From Endocrinology. 2001) (16)
Tabla II. Manifestaciones clínicas del MEN III (Tomado Endocrinology. 2001) (16)
La edad de presentación es muy variable y oscila entre los 10 meses y los 70 años con un pico de aparición en la tercera década de la vida (6). Los tumores solitarios parecen relacionarse con pacientes más jóvenes (7).
En relación al sexo, existen datos contradictorios, hay autores que opinan que es más frecuente en el sexo femenino (6) mientras que otros creen que lo es en el masculino (10), aunque, en cualquier caso, la diferencia entre sexos puede considerarse inapreciable (6).
Existe consenso en que la localización más frecuente es la piel. En cuanto a la localización intraoral hay discrepancia ya que algunos autores la consideran muy poco habitual (2-4) y otros no tan infrecuente (7). En este caso, la lesión se situaba en la mucosa yugal. Tanto ésta como la lengua se consideran los lugares de asiento más frecuentes a nivel intraoral (2-4,7). También se ha descrito, aunque en muy raras ocasiones, la localización central a nivel intraóseo, en la mandíbula (2-4,7).
Habitualmente se presentan como una masa única, nodular, bien definida, móvil y sesil. De crecimiento lento, suelen ser asintomáticas. Cuando originan algún síntoma, suele ser consecuencia de la compresión nerviosa y pueden producir dolor y parestesias (4,6).
No existe ninguna prueba de imagen diagnóstica. Sin embargo, la RNM proporciona más información sobre la extensión y límites del tumor que la TAC. Los neurofibromas se manifiestan como imágenes hipo ó isointensas en T1 e hiperintensas en T2 (Fig 1), bien delimitados y de consistencia homogénea (6).
Tampoco la PAAF, previa a la cirugía, suele ser diagnóstica en la mayoría de las ocasiones. El diagnóstico definitivo proviene del análisis histológico de la pieza quirúrgica (6). La imagen macroscópica es la de una tumoración de consistencia homogénea, coloración blanquecina y aspecto brillante (6). Microscópicamente suelen verse células fusiformes distribuidas irregularmente en un estroma compuesto por fibras de colágeno y acúmulos de material mucoide (6). Con tinciones de plata se pueden demostrar pequeños axones distribuidos en el tejido tumoral (7). Con las técnicas de inmunohistoquímica se aprecia positividad para la proteína S-100 (6,9,17).
El tratamiento electivo es la extirpación quirúrgica de las lesiones aisladas, intentando ser lo más conservador posible con el segmento del nervio que origina el tumor, aunque a menudo hay que sacrificar el nervio ya que este tumor crece invadiendo la estructura nerviosa de la cual deriva (4,6). Dado que hay descritos casos de regresión espontánea tras la pubertad, algunos autores recomiendan realizar el tratamiento después de la misma (2). El neurofibroma solitario no presenta prácticamente riesgo de malignización (12), en cambio, puede presentarse una degeneración sarcomatosa entre un 3 y un 15% cuando son múltiples o se asocian a la enfermedad de von Recklinghausen ó al síndrome poliglandular MEN III (4-6) y han sido sometidos a múltiples extirpaciones (3,5,6).
La recurrencia del neurofibroma solitario suele ser escasa (4), aunque hay autores que defienden una mayor tasa cuando se localiza en cabeza y cuello con respecto a otras localizaciones corporales (5,18).
BIBLIOGRAFÍA
1. Marx R, Stern D, eds. Oral and maxillofacial pathology. A rationale for diagnosis and treatment. Illinois: Quintessence Publishing Co, Inc; 2002. p. 418 [ Links ]
2. Grinspan D. Enfermedades de la boca. Semiología, patología, clínica y terapéutica de la mucosa bucal. Buenos Aires: Editorial Mundi S.A.I.C. y F.; 1976. p. 1910-1 [ Links ]
3. Sapp JP, Eversole LR, Wysocki GP, eds. Patología oral y maxilofacial contemporánea. Madrid: Harcourt Brace de España, S.A; 1998. p. 295-7 [ Links ]
4. Alatli C, Öner B, Ünür M, Erseven G. Solitary plexiform neurofibroma of the oral cavity. A case report. Int J Oral Maxillofac Surg 1996;25:379-80 [ Links ]
5. Wise J, Patel S, Shah J. Management issues in massive pediatric facial plexiform neurofibroma with neurofibromatosis type I. Head & Neck Feb 2002; 207-11 [ Links ]
6. Almela Cortés R, Faubel Serra M, Cueva Ruíz C, Conde Pérez de la Blanca I. Neurofibroma plexiforme de nervio facial intraparotídeo. Revisión de la literatura. Anales ORL Iber-Amer 2001;28:363-70 [ Links ]
7. Neville WB, Damm DD, Allen CM, Bouquot JE, eds. Oral & Maxillofacial Pathology. Philadelphia: WB Saunders company; 1995. p. 380-1 [ Links ]
8. Fauci A, Braunwald E, Isselbacher K, Wilson J, Martín J, Kasper D, et al, eds. Principios de Medicina Interna. Madrid: McGraw-Hill - Interamericana de España S.A.U.; 1998. p. 2738 [ Links ]
9. Fisher D, Chu P, McCalmont T. Solitary plexiform neurofibroma is not pathognomonic of von Recklinghausen's neurofibromatosis: a report of a case. Iternational Journal of Dermatology 1997;36:435-52 [ Links ]
10. Fitzpatrick T, Johson R, Wolff K, Polano M, Suurmond D, eds. Atlas de dermatología clínica. México: McGraw - Hill Interamericana editores, S.A. de C.V.; 1998. p. 458 [ Links ]
11. Weber AL, Montandon C, Robson CD. Neurogenic tumors of the neck. Radiol Clin North An 2000;38:1077-90 [ Links ]
12. Val-Bernal JF, Figols J, Vazquez - Barquero A. Cutaneous plexiform schwannoma associated with neurofibromatosis type 2. Cancer 1995;76: 1181-6 [ Links ]
13. Pujol RM, Matias - Guiu X, Miralles J, Colomer A, de Moragas JM. Multiple idiopathic mucosal neuromas: a minor form of multiple endocrine neoplasia type 2B or a new entity?. J Am Acad Dermatol 1997;37:349-52 [ Links ]
14. Gupta A, Cohen BH, Ruggieri P, Packer RJ, Phillips PC. Phase I study of thalidomide for the treatment of plexiform neurofibroma in neurofibromatosis 1. Neurology 2003;60:130-2 [ Links ]
15. Hernández G, Serrano MC, Baca R. Neurofibroma de la mucosa alveolar. A propósito de un caso. Medicina Oral 1998;3:107-13 [ Links ]
16. De Groot LJ, Jameson JL, Burger HG, Loriaux DL, Marshall JC, Melmed S, et al, eds. Edocrinology. Philadelphia: W.B. Saunders Company.; 2001. p. 2518 [ Links ]
17. Nagasaka T, Lai R, Sone M, Nakashima T, Nakashima N. Glandular malignant peripheral nerve sheath tumor: an unusual case showing histologically malignant glands. Arch Pathol Lab Med 2000;124:1364-8 [ Links ]
18. Needle MN, Cnaan A, Dattilo J, Chatten J, Phillips PC, Shochat S, et al. Prognostic signs in the surgical management of plexiform neurofibroma: the Children's Hospital of Philadelphia experience, 1974-1994. J Pediatr 1997; 131:678-82 [ Links ]

