










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Ed. impresa)
versión impresa ISSN 1698-4447
Med. oral patol. oral cir. bucal (Ed.impr.) vol.10 no.5 nov./dic. 2005
Poliposis familiar hereditaria y síndrome de Gardner: Aportación de la exploración odontoestomatológica a su diagnóstico y descripción de un caso
Hereditary familial polyposis and Gardner's syndrome: Contribution of the odonto-stomatology examination in its diagnosis and a case description
Eduardo Chimenos-Küstner (1), Montserrat Pascual (2), Ignacio Blanco (3), Fernando Finestres (4)
(1) Estomatólogo. Doctor en Medicina y Cirugía. Profesor Titular de Medicina Bucal,
Facultad de Odontología, Universidad de Barcelona
(2) Estomatóloga. Licenciada en Medicina y Cirugía. Área Básica de Salud Sant Roc (Badalona).
Profesora Asociada de la Facultad de Medicina de la Universidad Autónoma de Barcelona
(3) Oncólogo. Doctor en Medicina y Cirugía. Unidad de Consejo Genético, Servicio de Prevención y Control del Cáncer,
Instituto Catalán de Oncología. Profesor Asociado de Biología Celular y Anatomía Patológica,
Facultad de Medicina, Universidad de Barcelona
(4)Radiólogo y Estomatólogo. Doctor en Medicina y Cirugía. Profesor Asociado de Medicina Bucal,
Facultad de Odontología, Universidad de Barcelona
Correspondencia:
Dr. Eduardo Chimenos Küstner
Vía Augusta 124, 1º 3ª
08006 - Barcelona (España)
E-mail:
Recibido: 23-05-2004 Aceptado: 20-11-2004
Chimenos-Küstner E, Pascual M, Blanco I, Finestres F. Hereditary familial polyposis and Gardner's syndrome: Contribution of the odonto-stomatology examination in its diagnosis and a case description. Med Oral Patol Oral Cir Bucal 2005;10:402-9.
© Medicina Oral S. L. C.I.F. B 96689336 - ISSN 1698-4447
RESUMEN
La poliposis adenomatosa familiar (PAF) y su variante fenotípica, el síndrome de Gardner, constituyen una infrecuente patología hereditaria autosómica dominante. Se caracterizan por el desarrollo, generalmente durante la segunda y tercera década de la vida, de múltiples pólipos adenomatosos en el colon y en el recto. Estos pólipos tienen un riesgo elevado de transformación maligna subsiguiente, cosa que suele ocurrir en las décadas tercera y cuarta de la vida. Las manifestaciones fenotípicas de la PAF pueden ser muy variadas. Así, además de los pólipos colorrectales, los individuos afectos pueden presentar manifestaciones extracolónicas, entre las que se destacan: pólipos gastroduodenales, quistes dermoides y epidermoides, tumores desmoides, hipertrofia congénita del epitelio pigmentario de la retina, alteraciones óseas en los maxilares y en el esqueleto y anomalías dentarias. En este trabajo se revisan los aspectos más importantes del complejo, mostrando un ejemplo del mismo en base a un caso clínico bien documentado. Cabe destacar la importancia de las exploraciones odontoestomatológicas, entre otras, como medio para alcanzar el diagnóstico de presunción, cuya confirmación es vital para el enfermo.
Palabras clave: Poliposis familiar hereditaria, síndrome de Gardner, osteomas, marcadores diagnósticos.
ABSTRACT
Familial adenomatous polyposis (FAP) and its phenotype variant, Gardner's syndrome, constitute a rare autosomal dominant inherited disorder. They are characterised by the development, generally during the second and third decades of life, of multiple adenomatous polyps in the colon and rectum. These polyps have a high risk of subsequently becoming malignant, which normally occurs in the third and fourth decades of life. The phenotypical features of FAP can be very variable. As well as colorectal polyps, these individuals can present with extra-colonic symptoms, among which are particularly: gastro-duodenal polyps, dermoid and epidermoid cysts, desmoid tumours, congenital hypertrophy of the retinal pigment epithelium, disorders of the maxillary and skeletal bones and dental anomalies. In this paper the most important aspects of this syndrome are reviewed, showing an example based on a well documented clinical case. The importance of odonto-stomatological examinations should be pointed out, among others, as a means of reaching a presumptive diagnosis, whose confirmation is vital to the patient.
Key words: Hereditary familial polyposis, Gardner's syndrome, osteomas, diagnostic markers.
INTRODUCCIÓN
La poliposis adenomatosa familiar (PAF, en inglés Familial Adenomatous Polyposis - FAP) es una infrecuente enfermedad hereditaria autosómica dominante, que se caracteriza por el desarrollo, generalmente durante la segunda y tercera década de la vida, de múltiples pólipos adenomatosos (número mayor de 100) en el colon y en el recto. Estos pólipos tienen un riesgo elevado de transformación maligna subsiguiente, cosa que suele ocurrir en las décadas tercera y cuarta de la vida. Las manifestaciones fenotípicas de la PAF pueden ser muy variadas. Así, además de los pólipos colorrectales, los individuos afectos pueden presentar manifestaciones extracolónicas, entre las que se destacan: pólipos gastroduodenales, quistes dermoides y epidermoides, tumores desmoides, hipertrofia congénita del epitelio pigmentario de la retina (HCEPR), alteraciones óseas en los maxilares y en el esqueleto y anomalías dentarias (1). El Síndrome de Gardner se caracteriza por la asociación de pólipos colorrectales, quistes epidérmicos cutáneos y osteomas mandibulares y de huesos largos. Se considera que el Síndrome de Gardner es una variante fenotípica de la PAF. En la actualidad se conoce la base genética de la Poliposis Adenomatosa Familiar y sus diferentes variantes fenotípicas. La PAF y el síndrome de Gardner son debidos a la presencia de mutaciones en línea germinal en el gen APC (Adenomatous Polyposis Coli gene), localizado en el brazo largo del cromosoma 5 (2). Empieza a conocerse la correlación fenotipo-genotipo en la PAF, de forma que determinadas manifestaciones fenotípicas se relacionan con mutaciones en áreas concretas del gen APC. Las manifestaciones extracolónicas en la PAF (por ejemplo osteomas, quistes dermoides, etc.) se relacionan con mutaciones localizadas entre los codones 1395 y 1578 (2). Esta correlación fenotipo-genotipo tendrá una importancia vital cuando sea posible instaurar técnicas de reparación genómica dirigida (terapia génica). En la actualidad se utiliza para determinar las medidas de cribado más adecuadas para cada caso. En ocasiones no es posible identificar una mutación patogénica en el gen APC (3).
Es objetivo de este trabajo destacar la importancia que tiene una detección precoz de dicha patología, teniendo en cuenta que el hallazgo de lesiones que asientan en los maxilares desempeña un papel relevante de carácter diagnóstico.
MANIFESTACIONES CLÍNICAS
Osteomas. Los osteomas son lesiones osteogénicas benignas caracterizadas por proliferación lenta de hueso compacto o medular. Pueden ser centrales, periféricos o extraesqueléticos. Los osteomas centrales proceden del endostio, los periféricos del periostio y los extraesqueléticos se desarrollan en tejidos blandos, como un músculo. La lesión puede suceder en más de un hueso o con más de un osteoma en un único hueso. En la región maxilofacial, el tipo perióstico puede aparecer tanto externamente como en los senos paranasales. Es más frecuente en los senos frontal y etmoideo que en los senos maxilares. Estructuralmente los osteomas pueden dividirse en tres tipos, según estén compuestos por hueso compacto, por hueso medular o por una combinación de hueso compacto y medular. Clínicamente, el osteoma periférico suele ser asintomático, pero puede producir tumefacción y causar asimetría. Desde un punto de vista radiográfico, la lesión se define como una radiopacidad bien circunscrita. La tomografía computerizada es la mejor técnica de imagen para el diagnóstico de un osteoma. Histológicamente, los osteomas suelen estar formados por trabéculas de hueso laminar con médula fibroadiposa. En el caso de enostosis, éstas consisten en islotes compactos de hueso laminar maduro (4-6). Si bien en la población general no son comunes los osteomas en los huesos faciales y en el cráneo, en los pacientes afectos de síndrome de Gardner sí lo son. Con frecuencia se trata de masas grandes, multilobuladas, en la región goníaca. Muchas de ellas pueden confluir con osteomas adyacentes. Algunos osteomas pueden adoptar forma de gota, que parece colgar del borde inferior del borde mandibular o del cóndilo. Los osteomas que se originan en la médula ósea remedan enostosis. Aproximadamente el 50 % de los casos presentan tres o más osteomas en los maxilares, además de otras localizaciones. Un asiento frecuente de este tipo de exostosis es el hueso frontal. Los osteomas procedentes de hueso endocondral son raros, incluso en este síndrome. Cuando se encuentran en huesos largos, como la tibia o el fémur, adoptan más el aspecto de un engrosamiento cortical, que el de un osteoma verdadero. Es muy importante tener en cuenta que la aparición de osteomas precede las otras manifestaciones de este síndrome, incluida la poliposis intestinal. También debe recordarse que, con una incidencia del 17 %, pueden encontrarse odontomas, dientes supernumerarios y dientes impactados (4-8).
Poliposis intestinal. La poliposis adenomatosa familiar del colon es el síndrome más frecuente entre las poliposis hereditarias (1/8000). La mayoría de los individuos tienen una historia familiar de esta patología, pero hasta un 30 % de los pacientes pueden presentar una nueva mutación dominante (mutación "de novo") y ser el primer miembro afecto de su familia. Los pólipos suelen desarrollarse con posterioridad a los osteomas. La mayoría aparecen durante la segunda y tercera décadas de la vida. Su transformación maligna es un dato constante, que sólo depende del tiempo. En la pubertad, la tasa de malignización es del 5 %, con un incremento que alcanza el 50 % a los 30 años y el 100 % de los casos en pacientes de más de 50 años de edad. Además, el síndrome de Gardner se asocia con la presencia de pólipos en cualquier tramo del tubo digestivo, que, a su vez, pueden malignizarse. Se ha descrito también la presencia de tumores malignos en otras localizaciones: carcinomas en la ampolla de Vater, meduloblastomas, carcinomas de tiroides y hepatoblastomas (4,9,10). La prevalencia del cáncer en pacientes con poliposis adenomatosa familiar sintomática oscila entre el 47 y el 67%. Los registros demuestran que a pesar del conocimiento de la enfermedad, el 59 % de estos pacientes fallecen por la extensión metastásica de un cáncer colorrectal. Sin embargo, cuando se estudia a familiares asintomáticos que presentan el fenotipo PAF, la prevalencia de transformación maligna se observa sólo en el 2 %. El impacto que puede tener la detección asintomática sobre la supervivencia es muy notable. Los pacientes detectados en etapa sintomática tienen una supervivencia a los 5 años del 40 %, en comparación con el 93 % de los individuos asintomáticos detectados mediante programas de detección (11,12). Desde el punto de vista histológico, los pólipos de este síndrome son adenomatosos y asientan sobre todo en el colon. Sin embargo, el intestino delgado, en particular el duodeno, puede afectarse en este proceso. También se observan con frecuencia pólipos gástricos (pólipos de las glándulas fúndicas), de naturaleza hamartomatosa. Los adenomas son tubulares, vellosos o una combinación de ambos. Si bien los adenomas de colon, en especial los adenomas vellosos, pueden malignizar en la población general, debe tenerse presente que en los pacientes con síndrome de Gardner esto es inevitable y por lo general ocurre de forma precoz. Si no se practica una colectomía, se desarrollará un adenocarcinoma. Estos tumores pueden secretar cantidades variables de mucina, pero el pronóstico depende más de la extensión del tumor por la pared intestinal, que de las características histológicas específicas (4).
Quistes epidermoides, dermoides y sebáceos. Los quistes sebáceos se desarrollan en un 60 % de casos, aproximadamente. En promedio, el número de quistes es de 4, si bien algunos individuos desarrollan 20 o más. Los quistes se observan con mayor frecuencia en la cara, el cuero cabelludo, los brazos y las piernas. También suelen aparecer antes de la pubertad y de que se manifieste la poliposis intestinal. La estructura de su pared es análoga a la de la piel y suele contener materias organizadas, como grasa, pelos, glándulas, etc. Están delimitados por un epitelio escamoso estratificado delgado, productor de queratina. (4,10).
Fibromas y fibromatosis en tejidos blandos. Estos tumores de tejidos blandos, a menudo denominados desmoides abdominales o extraabdominales, son masas fibrosas infiltrantes, que se observan en el 15 al 30 % de los casos. Tales tumores fibrosos se observan sólo en un 5 % de los casos de PAF, por su menor penetrancia en el gen SG-PAF. Algunos de estos tumores aparecen de novo, otros después de un tratamiento quirúrgico (sobre todo cirugía abdominal) y otros después de extirpar tumores desmoides previos. Los tumores localizados en el área maxilofacial se ha observado que infiltran la musculatura masticatoria y suprahioidea (4,10).
Hipertrofia congénita del epitelio pigmentario de la retina (HCEPR). Hasta un 75 % de los pacientes con poliposis familiar adenomatosa (PFA) presentan una hipertrofia congénita del epitelio pigmentario de la retina, lo que se detecta con facilidad mediante oftalmoscopia. Como no es frecuente que un individuo normal presente este tipo de lesiones, su detección, junto a algunos de los datos clínicos ya mencionados, debe hacer pensar en la posibilidad de un síndrome de Gardner (10).
DIAGNÓSTICO DIFERENCIAL
El síndrome de Gardner con total expresión será evidente desde las observaciones clínicas y radiográficas. Algunas entidades que pueden sugerir masas radiodensas múltiples u odontomas con dientes impactados y supernumerarios en la ortopantomografía son la displasia cleidocraneal, la displasia oseocementaria florida y la displasia oseocementaria periapical. Diversas enfermedades que producen poliposis intestinal incluyen la poliposis colónica juvenil, el síndrome de Turcot (caracterizado por la asociación de pólipos en colon y recto y tumores malignos del sistema nervioso central - gliomas-), el síndrome de Cowden (hamartomatosis múltiple de herencia autosómica dominante, consistente en triquilemomas que se originan en células del folículo piloso) y el síndrome de Peutz-Jeghers (poliposis gastrointestinal hemorragípara y manchas melánicas en labios y mucosa oral; herencia autosómica dominante), entre muchos otros. (Si bien la mayoría de los pólipos que aparecen en el síndrome de Peutz-Jeghers se localizan en el intestino delgado, algunos aparecen también en el intestino grueso). No obstante, tan sólo el síndrome de Gardner presenta todos o la mayoría de trastornos referidos en la tétrada de osteomas, tumores fibrosos, quistes sebáceos y poliposis intestinal (4,5).
DIAGNÓSTICO
Para establecer el diagnóstico, debe explorarse al paciente de forma minuciosa. Habrá que averiguar si existen quistes sebáceos y masas correspondientes a tumores fibrosos. Se recomienda la realización de radiografías de cráneo y panorámica (ortopantomografía). Si se observan lesiones óseas radiopacas u osteomas, es conveniente biopsiar al menos una de ellas, para confirmar su identidad. Deben solicitarse pruebas como tránsito gastrointestinal con papilla de bario y enema opaco, para estudiar el intestino distal. Cualquier hallazgo sospechoso o la detección de un síndrome de Gardner establecido exigen la práctica de una colonoscopia (4,7). Se recomienda el estudio genético del gen APC en todo individuo con sospecha de PAF o Síndrome de Gardner.
TRATAMIENTO
No existe un tratamiento etiológico de la PAF ni del Síndrome de Gardner, siendo el único disponible el sintomático. Dado que prácticamente el 100% de los pacientes afectos de PAF pueden desarrollar un cáncer colorrectal, el diagnóstico de síndrome de Gardner suele requerir una colectomía profiláctica. En algunos casos se realizan estudios seriados con colonoscopias, para retrasar la colectomía, pero estos estudios conllevan un riesgo de dejar sin detectar una transformación maligna. Si bien los osteomas no requieren ser extirpados, a menudo se eliminan, debido a su apariencia y a la interferencia que suponen a la movilidad. Dada la dirección genética del síndrome, al cabo de varios meses o años pueden aparecer nuevos osteomas. Los quistes sebáceos se extirpan a demanda del paciente. Los tumores fibrosos y las fibromatosis suelen escindirse, pero su capacidad de infiltración local exige crioterapia y extirpación en bloque, con un margen de seguridad de 1 cm (4).
PRONOSTICO Y SEGUIMIENTO
En los pacientes con síndrome de Gardner que se diagnostican de forma precoz y a los que se practica una colectomía, el pronóstico suele ser muy bueno. Tienden a llevar una vida casi normal. Sin embargo, el seguimiento debe realizarse con una periodicidad de 6 - 12 meses, tanto en el caso de los pacientes, como de sus familiares, dado que la herencia tiene carácter autosómico dominante (4). Se ha observado que el estudio radiográfico maxilofacial (ortopantomografía, tomografía computerizada) puede ser de gran utilidad para el diagnóstico precoz de esta patología y la instauración temprana del tratamiento más oportuno y seguimiento del paciente y sus familiares (7,8), si bien sólo la existencia concomitante de poliposis intestinal permite el diagnóstico definitivo del síndrome (10). Una vez realizado el diagnóstico clínico, la última palabra la tendrá el estudio genético, aunque éste también tiene sus limitaciones (3).
CASO CLINICO
Paciente de sexo femenino, de 42 años de edad, que acude a la consulta por presentar un dolor inespecífico en la hemiarcada inferior derecha (cuarto cuadrante), de varios días de evolución. En la exploración clínica se constata dolor a la percusión del primer molar -4.6-. La paciente explica haber sufrido algunos episodios dolorosos desde hace aproximadamente 1 año, que se resolvieron de forma espontánea. La palpación de las estructuras blandas y duras del área maxilofacial pone de manifiesto la presencia de protuberancias de consistencia ósea, compatibles con exostosis, de unos 5 mm de diámetro, en la hemiarcada superior izquierda (segundo cuadrante, a la altura del fondo del vestíbulo del primer premolar), así como en la hemiarcada inferior izquierda (tercer cuadrante), cerca del ángulo mandibular, que responde a la descripción mencionada anteriormente: "en forma de gota, a nivel del borde inferior del gonión".
Al efectuar una radiografía periapical de la zona dolorosa (molar 4.6), se observaron sendas imágenes radioopacas de aspecto algodonoso. Una de ellas se extiende desde mesial de la raíz del 4.7 hacia el espacio comprendido entre los ápices radiculares del 4.6, aproximándose al conducto dentario, hasta alcanzar la altura de la raíz del ápice mesial de este molar. La otra, con asentamiento más coronal, ocupa parte del espacio comprendido entre las raíces de los dientes 4.6 y 4.5 (figura 1).
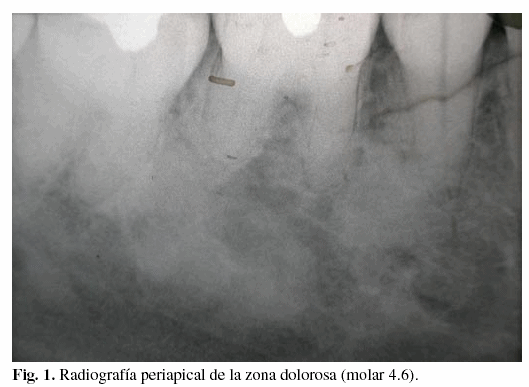
En el interrogatorio, la paciente relata estar afecta de poliposis familiar adenomatosa, variante síndrome de Gardner, diagnosticada en la pubertad. Fue intervenida quirúrgicamente a los 21 años, practicándose colectomía total con anastomosis ileorrectal. Presenta también quistes epidermoides en la espalda y pólipos gástricos. El estudio genético de la paciente permitió identificar la existencia de una mutación en el exón 9 del gen APC que provoca un cambio de aminoácido de triptófano a codón de terminación (W421X), generando una proteína truncada. Esta alteración genética es la responsable de la Poliposis Adeno matosa Familiar descrita en esa familia. El estudio genético ha permitido identificar que sus dos hijos son portadores de la misma mutación genética.
Se realiza un estudio radiográfico de los tres miembros portadores (madre y los dos hijos). En la ortopantomografía de la paciente, se observan diversas imágenes radioopacas bien delimitadas, de tamaños variables y características similares a las descritas en la proyección intraoral periapical, que se extienden por toda la mandíbula (figura 2).

En una ortopantomografía de la hija, se constata la presencia de múltiples lesiones radiodensas mandibulares. En la ortopantomografía actual del hijo, sometido a tratamiento ortodóncico, se observa la inclusión dentaria del canino 2.3.
La historia de esta familia es amplia, como demuestra el esquema mostrado, correspondiente a 1999 (figura 3). Puede comprobarse cómo en esta familia, a pesar de los casos clínicos evidentes y de haberse identificado la mutación responsable de la enfermedad, hay muchos miembros que no sólo no han accedido al estudio genético para conocer si son o no portadores de la mutación genética en la familia, sino que ni tan sólo han accedido a realizarse el cribado de la enfermedad mediante colonoscopias.
En estas familias, los hallazgos fenotípicos extracolónicos, como son los osteomas, pueden permitir identificar a los individuos con un riesgo elevado de haber heredado la enfermedad y, por lo tanto, reforzar la necesidad del inicio de las pruebas de cribado.
DISCUSION
La importancia de la patología descrita radica en el gran potencial de malignización que presentan las lesiones polipoides intestinales. El ejemplo clínico expuesto pretende ilustrar una situación, en la que diversas especialidades médicas pueden aportar datos y participar en el diagnóstico precoz. Éste, por su parte, permitirá llevar a cabo un tratamiento preventivo o en fases incipientes de la enfermedad, de manera que contribuirá a mejorar el pronóstico.
Las lesiones óseas condensantes, en forma de osteomas o de enostosis, pueden plantear problemas de diagnóstico diferencial con diversas entidades nosológicas, como las que ya se han indicado con anterioridad (13). Sin embargo, la relativa facilidad con que se detectan clínica y radiológicamente, constituye un instrumento muy útil para el diagnóstico precoz. Odontólogos y estomatólogos tienen mucho que aportar en este sentido, amén de todos los especialistas en técnicas de diagnóstico por la imagen. Vale la pena destacar trabajos como los realizados por Thakker et al., en 1995 (7), o Herrmann et al., en 2003 (10), quienes tienen en cuenta signos extradigestivos, como los osteomas, como elementos contribuyentes al diagnóstico precoz de la PAF.
También las lesiones cutáneas y oculares tienen un fácil acceso exploratorio, de modo que el campo de los dermatólogos, de los oftalmólogos y de otros profesionales de la salud puede contribuir asimismo a su diagnóstico precoz.
Algo más difícil resulta la exploración del aparato digestivo, por cuanto las técnicas exploratorias pueden parecerle más molestas al paciente y no siempre se va a dejar explorar de forma conveniente, al igual que ocurre con el estudio genético. Éstas son, sin embargo, las investigaciones más importantes para el control de la mencionada patología.
En nuestra mano, como profesionales de la salud que somos, está, por tanto, saber orientar de forma adecuada al paciente, dirigiéndolo a los profesionales más indicados en cada caso.
BIBLIOGRAFIA
1. Online Mendelian Inheritance in Man. OMIM (TM 6 / 7 / 2001). John Hopkins University: Baltimore, MD. MIM Number: *175100: World Wide Web URL: http://www.ncbi.nlm.nih.gow/omim/ [ Links ]
2. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Human Molecular Genetics 2001; 10: 721-33. [ Links ]
3. Bisgaard ML, Ripa R, Knudsen AL, Bulow S. Familial adenomatous polyposis patients without an identified APC germline mutation have a severe phenotype. Gut 2004; 53: 266-7. [ Links ]
4. Marx RE, Stern D. Gardner Syndrome. En: Oral and Maxillofacial Pathology. A Rationale for Diagnosis and Treatment. Chicago: Quintessence Publishing Co, Inc.; 2003. p. 776-8. [ Links ]
5. Matteson SR. Tumores benignos de los maxilares. En: White SC, Pharoah MJ. Radiología oral. Principios e interpretación. Madrid: Elsevier Science; 2002. p. 378-419. [ Links ]
6. Sayan NB, Üçok C, Karasu HA, Günhan Ö. Peripheral Osteoma of the oral and maxillofacial region: a study of 35 new cases. J Oral Maxillofac Surg 2002; 60: 1299-301. [ Links ]
7. Thakker N, Davies R, Horner K, Armstrong J, Clancy T, Guy S et al. The dental phenotype in familial adenomatous polyposis: diagnostic application of a weigheted scoring system for changes on dental panoramic radiographs. J Med Genetics 1995; 32: 458-64. [ Links ]
8. Aggarwal VR, Sloan P, Horner K, Macfarlane TV, Clancy T, Evans G et al. Dento-osseous changes as diagnostic markers in familial adenomatous polyposis families. Oral Diseases 2003; 9: 29-33. [ Links ]
9. Payne M, Anderson JA, Cook J. Gardner's syndrome - a case report. Br Dent J 2002; 193: 383-4. [ Links ]
10. Herrmann SM, Adler YD, Schmidt-Petersen K, Nicaud V, Morrison C, Paul M et al. The concomitant occurrence of multiple epidermal cysts, osteomas and thyroid gland nodules is not diagnostic for Gardner syndrome in the absence of intestinal polyposis: a clinical and genetic report. Br J Dermatol 2003; 149: 877-83. [ Links ]
11. Galle TS, Juel K, Bulow S. Scand. J Gastroenterol 1999; 34: 808-12. [ Links ]
12. Cervantes Ruipérez A: Poliposis adenomatosa familiar. En: Rodés Teixidor J, Guardia Massó J (eds.). Medicina Interna (tomo I). Barcelona: Masson; 1997. p. 1399-1400. [ Links ]
13. White SC, Pharoah MJ. Oral radiology. Principles and interpretation. St Louis, Missouri: Mosby; 2004. p. 410-57. [ Links ]

