










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Internet)
versión On-line ISSN 1698-6946
Med. oral patol. oral cir.bucal (Internet) vol.11 no.4 jul. 2006
Fibropapilomatosis oral múltiple como manifestación inicial de Síndrome de Cowden. Caso clínico
Multiple oral fibropapillomatosis as an initial manifestation of Cowden Syndrome. Case report
Luis Miguel Capitán Cañadas1, José Luis Salinas Sánchez2, Sergio Luis Martínez Castillo2,
Ildefonso Leopoldo Labrot Moleón1, David Durán Moreno1, Darío Sánchez López2, Eduardo Valencia Laseca3
1Médico Interno Residente
2Médico Adjunto
3Jefe de Servicio. Servicio de Cirugía Oral y Maxilofacial. Hospital Universitario Virgen de las Nieves. Granada
Dirección para correspondencia
RESUMEN
El síndrome de Cowden es una infrecuente enfermedad hereditaria englobada dentro de las poliposis gastrointestinales de tipo hamartomatoso. Se caracteriza por asociar anomalías cutaneomucosas y por la extraordinaria tendencia a desarrollar neoplasias malignas, principalmente de mama y tiroides. La importancia de un diagnóstico precoz del síndrome y de un adecuado screening tumoral en pacientes con lesiones papilomatosas cutaneomucosas, nos va a permitir adelantarnos en el diagnóstico de patologías con enorme morbimortalidad asociada en caso de una detección tardía. Presentamos el caso de una paciente diagnosticada de síndrome de Cowden tras consultar por lesiones papilomatosas labiales de largo tiempo de evolución y tratada posteriormente de cáncer de mama y riñón en estadios iniciales.
El correcto diagnóstico de una patología banal de mucosa oral, nos ha permitido actuar de manera muy precoz frente a la patología neoplásica asociada a dicha enfermedad.
Palabras clave: Síndrome de Cowden, hamartomas múltiples, oral, papiloma.
ABSTRACT
Cowden syndrome is a rare hereditary disease included within hamartoma-type gastrointestinal polyposis. It is characterised by associated mucocutaneous anomalies and by the extraordinary tendency to develop malignant neoplasia, mainly in the breast and thyroid. Early diagnosis of the syndrome and adequate tumoral screening in patients with mucocutaneous papillomatosis make it possible to make an earlier diagnosis of associated pathologies which have great morbidity when detected late. We present the case of a patient diagnosed with Cowden syndrome after consultation for labial papillomatous lesions of long evolution who was subsequently treated for breast and kidney cancer in initial stages.
The correct diagnosis of a banal pathology of oral mucosa made it possible for us to take early action against the neoplastic pathology associated with this disease.
Key words: Cowden syndrome, multiple hamartomas, oral, papilloma.
Introducción
Descrita inicialmente por Lloyd y Dennis en 1963 (1) el síndrome de Cowden o síndrome de los hamartomas múltiples, es una infrecuente genodermatosis hereditaria de transmisión autosómica dominante con expresividad variable (2), englobada dentro de la gran familia de las poliposis gastrointestinales hereditarias (3). Estas poliposis se caracterizan por el tipo de pólipo dominante: hamartomatoso o adenomatoso, estando los primeros definidos por un sobrecrecimiento de elementos celulares epiteliales y estromales de características normotípicas (2). Las poliposis hamartomatosas representan un pequeño pero no despreciable número de poliposis hereditarias con predisposición a degenerar en cáncer gastrointestinal (4).
La prevalencia del síndrome de Cowden es de 1:200000 (3) por lo que puede considerarse como enfermedad rara. (Código CIE-9-MC:759.6) (5). En el síndrome de Cowden encontramos hamartomas múltiples de origen endodérmico, ectodérmico y mesodérmico (3). Pese a tratarse de una enfermedad englobada en el área gastrointestinal, sus manifestaciones clínicas extraintestinales son manifiestas y en numerosas ocasiones condicionan el pronóstico del paciente (3).
- Manifestaciones gastrointestinales
Las manifestaciones del tracto gastrointestinal del síndrome de Cowden son prácticamente indistinguibles del síndrome de poliposis familiar juvenil (6). Se caracteriza por la presencia de múltiples pólipos hamartomatosos a nivel de todo el tracto gastrointestinal, localizados principalmente en colon y recto (4, 7), generalmente asintomáticos (8), lo que contribuye al infradiagnóstico de la enfermedad (4). Histológicamente se caracterizan por presentar un elevado número de glándulas normotípicas de contenido mucoide, inflamación y edema a nivel de la lámina propia e infiltrado linfoplasmocitario (9). Asimismo podemos encontrar ganglioneuromas y pólipos lipomatosos e inflamatorios (3). A nivel esofágico es posible encontrar acantosis glicogénica sin repercusión clínica asociada (10).
- Manifestaciones extraintestinales
Al contrario que en otras poliposis, las manifestaciones sistémicas del síndrome de Cowden son abundantes y suficientemente distintivas, lo que puede orientar su diagnóstico clínico (3). Las lesiones cutaneomucosas son las manifestaciones más destacadas del síndrome de Cowden; de hecho, aproximadamente el 80% de pacientes diagnosticados de síndrome de hamartomas múltiples, presenta o desarrollará en el futuro alguna manifestación de este tipo (11). Suelen ser lesiones de largo tiempo de evolución que, por norma general, se manifiestan en etapas iniciales de la infancia, antes del desarrollo de otros procesos neoplásicos asociados a la enfermedad, lo que pone de manifiesto la importancia de detectar a tiempo estas lesiones benignas e iniciar un adecuado screening tumoral de diagnóstico precoz (12). En cuanto al tipo de lesiones destacan triquilemomas faciales, queratosis acral, lipomas subcutáneos, queratosis palmoplantar, papilomas orales y labiales con un característico aspecto de empedrado (13). De manera mucho menos frecuente puede asociarse al síndrome de Cowden la aparición de vitíligo, neuromas, xantomas y manchas café con leche (14).
Pese a ser las manifestaciones clínicas más destacadas, las citadas lesiones pueden acompañarse de manifestaciones otorrinolaringológicas (15), oftalmológicas (16), ginecológicas (17), renales (18) y craneomaxilofaciales (3). Por su especial trascendencia destacamos el alto riesgo asociado de desarrollar patología neoplásica, principalmente de mama y tiroides (3), sin dejar de lado la posibilidad de desarrollar otro tipo de neoplasias (ovario, útero, riñón y meninges) (17).
Diagnóstico
Se basa fundamentalmente en criterios clínicos. En la actualidad se acepta el método diagnóstico de criterios clínicos mayores y menores propuesto por Salem y Steck (11), revisado y modificado posteriormente por el Internacional Cowden Consortium en el año 2000 (19) (Tabla 1 de Lesiones Patognomónicas y Criterios Mayores y Menores).
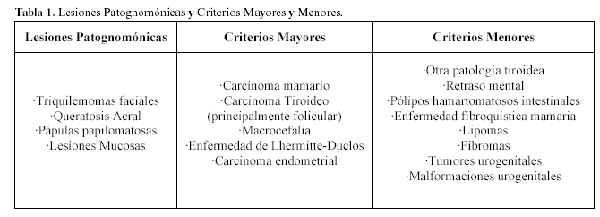
Se diagnosticará el síndrome de Cowden, cuando el paciente cumpla con alguno de los siguientes criterios:
1. Presencia de lesiones patognomónicas, siempre y cuando se dé alguna de las siguientes condiciones:
- Seis o más pápulas faciales, de las cuales tres o más deben ser triquilemomas.
- Pápulas faciales y papilomatosis en mucosa oral
- Papilomatosis en mucosa oral y queratosis acral.
- Seis o más lesiones queratósicas palmoplantares
2. Presencia de dos criterios mayores, uno de los cuales debe ser macrocefalia o enfermedad de Lhermitte Duclos.
3. Presencia de un criterio mayor y tres criterios menores.
4. Presencia de cuatro criterios menores.
Podremos diagnosticar de síndrome de Cowden a un familiar directo de un paciente afecto si cumple alguna de las siguiente condiciones (3):
1. Lesiones patognomónicas cutaneomucosas.
2. Algún criterio mayor con o sin criterio menor asociado.
3. Presencia de dos criterios menores.
Caso clínico
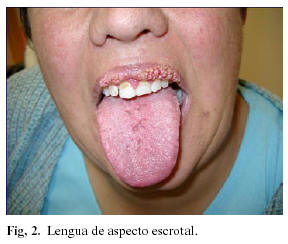
Paciente mujer de 42 años de edad, sin antecedentes personales de interés, que acude remitida por su médico de cabecera, a la consulta de Cirugía Oral y Maxilofacial del Hospital Universitario Virgen de las Nieves de Granada, por presentar múltiples lesiones de aspecto papilomatoso en labio superior (Figuras 1a y 1b) de aproximadamente quince años de evolución, según refiere la paciente. En la exploración intraoral de la paciente se puede apreciar asimismo la presencia de lengua de aspecto escrotal (Figura 2). Bajo anestesia local y en dos tiempos quirúrgicos, se procede a la exéresis completa de las lesiones. El estudio anatomopatológico de las mismas tipifica las lesiones como fibropapilomas.
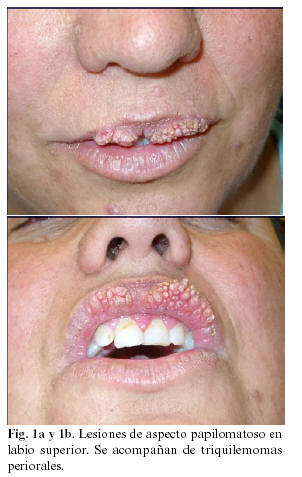
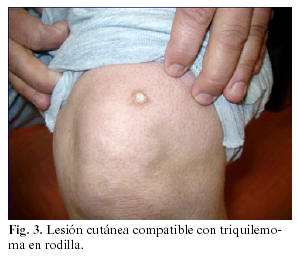
Por el característico aspecto empedrado de las lesiones mucosas, acompañadas en la exploración sistémica de triquilemomas cutáneos en regiones acrales (Figura 3), el largo tiempo de evolución de las mismas y la presencia de antecedentes familiares directos (madre fallecida de neoplasia de tiroides a los 49 años con presencia del mismo tipo de lesiones en mucosa labial, hermana con patología mamaria no filiada e hija intervenida de carcinoma papilar de tiroides) se decide derivar a la paciente al Servicio de Ginecología para despistaje de patología neoplásica mamaria, dada la fuerte sospecha, entre los diagnósticos diferenciales propuestos, de encontrarnos frente a un síndrome de Cowden.
En el Servicio de Ginecología de nuestro Hospital, la paciente es diagnosticada de patología fibroquística mamaria bilateral y de carcinoma ductal infiltrante grado GII de mama izquierda, que requiere tumorectomía más vaciamiento axilar homolateral y tratamiento complementario con quimio-radioterapia postoperatoria. Ante el diagnóstico clínico de genodermatosis de hamartomas múltiples (la paciente presenta una lesión patognomónica, un criterio mayor y tres criterios menores), se inicia un completo proceso de despistaje tumoral sistémico.
La paciente es valorada en el Servicio de Digestivo, donde se detecta la presencia de pólipos asintomáticos en mucosa rectal y colónica subsidiarios de seguimiento clínico; posteriormente es remitida al Servicio de Endocrinología para valoración de aumento de tamaño progresivo del tiroides asociado a hipertiroidismo clínico. Tras descartar malignidad mediante estudio citohistológico, la paciente es intervenida por el Servicio de Cirugía Endocrina practicándosele tiroidectomía total. El resultado anatomopatológico concluyente diagnostica la lesión como bocio coloide multinodular.
A los cinco meses de la cirugía tiroidea y tras la realización de una ecografía abdominopélvica de rutina, se detecta una masa en riñón derecho, que tras estudio con Tomografía Computerizada, presenta características radiológicas de malignidad. Derivada al Servicio de Urología, se decide practicar nefrectomía radical derecha más linfadenectomía supracava. El estudio histológico del espécimen quirúrgico diagnostica la lesión como carcinoma renal de células claras y granulares de componente fundamentalmente tubular. No precisa tratamientos complementarios postquirúrgicos.
En la actualidad la paciente presenta disminución progresiva de agudeza visual no filiada en estudio por parte del Servicio de Oftalmología.
Todos los procesos neoplásicos diagnosticados y tratados fueron detectados en estadios iniciales. Por el momento la paciente se encuentra en remisión completa de toda la patología tumoral diagnosticada y tratada. Pese a tener casi desde el principio un claro diagnóstico clínico, la paciente ha sido estudiada en el Centro Nacional de Investigaciones Oncológicas [Departamento de Genética Humana] confirmándose de manera definitiva que es portadora de la mutación c1093lnaGGAT en el gen PTEN, lo que ratifica el diagnóstico genético de Síndrome de Cowden. El estudio se ha ampliado al resto de familiares de primer grado para efectuar un adecuado despistaje e iniciar un estrecho seguimiento clínico en caso necesario.
Discusión
El síndrome de los hamartomas múltiples o síndrome de Cowden aparece fundamentalmente en caucasianos (95%) y afecta con mayor frecuencia a mujeres (60%) (12). Los síntomas suelen manifestarse entre la tercera y cuarta década de la vida (6). La mayoría de nuevos pacientes diagnosticados se presentan de manera aislada sin presentar historia de afectación familiar (3). Carlson et al (9) determinaron que únicamente un 10-15% de pacientes tenían algún familiar afectado. Posiblemente debido al infradiagnóstico de la enfermedad, la verdadera proporción entre casos esporádicos y familiares no se encuentra bien determinada (20). Aproximadamente un 80% de los pacientes es portador de mutaciones en el gen supresor de tumores PTEN [phosphatase and tensin homolog] localizado en el cromosoma 10q23.3 (21). Dicho gen inhibe el crecimiento tumoral actuando como regulador del crecimiento celular potenciado por la tirosínkinasa (10). Hasta la fecha no se han identificado otras mutaciones genéticas asociadas al síndrome (3). Las manifestaciones extraintestinales cutaneomucosas constituyen las características clínicas más relevantes de este síndrome (3). De hecho los cuatro criterios patognomónicos se encuentran relacionados con lesiones de este tipo (19). En numerosas ocasiones, como el caso que presentamos, son este tipo de lesiones las que pueden poner al especialista sobre la pista de un cuadro clínico mucho más complejo (3). Algo más del 80% de pacientes diagnosticados de síndrome de Cowden presentan manifestaciones dermatológicas (11). Las lesiones cutáneas se presentan como pápulas papilomatosas o liquenoides distribuidas por el territorio maxilofacial, cuello, dorso de las manos y antebrazos principalmente (22). En el territorio maxilofacial se concentran alrededor de la boca, orejas, periórbita y zona glabelar (23). Histopatológicamente estas lesiones, identificadas como triquilemomas, son de naturaleza hamartomatosa, derivan de las folículos pilosos y constituyen una de las manifestaciones sistémicas más significativas de la enfermedad (24, 25). La queratosis acral es la segunda manifestación dermatológica más frecuente (3), con la presencia de pápulas de superficie lisa o rugosa en el dorso de manos y pies. Se han descrito otras manifestaciones cutáneas menos frecuentes como vitíligo, manchas café con leche y melanosis (14).
En mucosa oral podemos encontrar lesiones papulosas fibromatosas; pese a que las localizaciones más frecuentes son labios, lengua, encía, reborde alveolar y mucosa yugal (26), se han descrito otras regiones afectas tales como paladar, úvula, orofaringe, laringe y mucosa nasal (22).
En pacientes con prótesis mucosoportada podemos apreciar componente irritativo lesional (27). Microscópicamente son lesiones poco específicas, con epitelio escamoso estratificado y numerosas bandas fibrosas (14). Es importante hacer un correcto diagnóstico diferencial de las lesiones fibropapilomatosas de mucosa oral que pueda ser de utilidad en el manejo del paciente (Tabla 2. Diagnóstico diferencial de lesiones fibropapilomatosas de mucosa oral).

Es posible observar asociado al síndrome la presencia de lengua escrotal (23), paladar ojival (3) y tendencia a la aparición de enfermedad periodontal (28).
Las lesiones benignas cutáneas y mucosas descritas suelen manifestarse en la infancia de manera previa al desarrollo de manifestaciones clínicas de mayor severidad asociadas al síndrome de Cowden (3), por lo que el reconocimiento precoz de este tipo de lesiones es crucial para el diagnóstico de la enfermedad y el inicio de un adecuado screening tumoral (16).
Entre las principales manifestaciones sistémicas extraintestinales asociadas al síndrome de los hamartomas múltiples destacan la posibilidad de desarrollar patología tiroidea y mamaria, principalmente de estirpe tumoral (3). Alrededor del 67% de pacientes diagnósticados de síndrome de Cowden presentan patología tiroidea, que es la manifestación sistémica más frecuente asociada al síndrome (29). Los principales cuadros tiroideos asociados descritos, han sido bocio coloide, adenoma folicular, carcinoma papilar y folicular, oncocitoma y quistes del conducto tirogloso (3). El paciente puede presentar hipo, hiper o eutiroidismo, sin existir una clara tendencia hacia ninguna de estas formas clínicas (29). Hasta un 3% de pacientes pueden desarrollar tiroiditis y hasta un 12% carcinoma de tiroides (30).
Algo más del 50% de los pacientes afectos de Cowden pueden desarrollar patología mamaria (16). La patología mamaria benigna asociada puede incluir formas como mastopatía fibroquística, fibroadenomas y malformaciones areolares (3). Dentro de las formas malignas destaca el carcinoma de mama (31), que es posiblemente la manifestación más preocupante del síndrome de Cowden y que puede llegar a afectar hasta a un 36% de los pacientes (11), lo que hace preciso un seguimiento estrecho del paciente, exploraciones clínicas periódicas, estudios mamográficos y un adecuado despistaje familiar.
A nivel genitourinario pueden asociarse menstruaciones irregulares, quistes ováricos y patología tumoral ovárica, de cérvix, endometrial y renal (17).
Pese a tratarse de una enfermedad originariamente digestiva y al contrario que ocurre en otras polipomatosis hamartomatosas, no está documentado el riesgo incrementado de desarrollar neoplasias intestinales (9, 16). No obstante, y hasta el momento en el que dispongamos de mayor información al respecto, es necesario un adecuado screening de despistaje tumoral gastrointestinal.
En aproximadamente un tercio de pacientes con síndrome de Cowden pueden aparecer alteraciones esqueléticas que incluyen craneomegalia, pectus excavatum, anomalías vertebrales y paladar ojival (32). Por último, existe una serie de cuadros que pueden desarrollarse en el contexto de síndrome de Cowden, alteraciones oculares como hipertelorismo, opacidades corneales y glaucoma (16), anomalías del sistema nervioso que pueden incluir neuromas de terminales nerviosos cutáneos, neurofibromas, enfermedad de Lhermitte Duclos y disminución progresiva de audición (15), patologías cardiorrespiratorias entre las que destacamos hipertensión arterial, defectos septales interauriculares, prolapso de válvula mitral, insuficiencia de válvula aórtica y mitral y hamartomas pulmonares (16). Otra miscelánea de tumores descritos (17, 18) en el contexto de síndrome de Cowden incluyen lipomas, liposarcomas, tumores meníngeos, linfomas no Hodgkin y carcinoma de células de Merkel.
Conclusión
A partir del diagnóstico diferencial de lesiones orales inespecíficas de largo tiempo de evolución se diagnosticó un cuadro clínico que también asociaba bocio tiroideo y neoplasias malignas de mama y riñón.
El síndrome de Cowden es una causa poco frecuente de lesiones fibropapilomatosas de mucosa oral y piel. El desarrollo de estas lesiones suele preceder a la instauración casi sistemática de patología tumoral, principalmente de mama y tiroides. Un adecuado diagnóstico precoz de las mismas va a facilitar el seguimiento, diagnóstico y tratamiento de estos pacientes en estadios iniciales, así como la realización de un screening familiar sistemático.
Bibliografía
1. Lloyd K, Dennis M. Cowdens disease: a possible new symptom complex with multiple system involvement. Ann Int Med 1963;58:136-42. [ Links ]
2. Boardman LA. Hereditable colon cancer syndromes: Recognition and preventive management. Gastroenterol Clin N Am 2002;31:1107-31. [ Links ]
3. Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: A clinical and molecular review. Am J Gastroenterol 2005;100:476-90. [ Links ]
4. Longy M, Lacombe D. Cowdens disease: report of a family and review. Ann Genet 1996;39:35-42. [ Links ]
5. Ramírez Díaz-Bernardo, J. Introducción. En: Izquierdo Martínez M, Avellaneda Fernández A, eds. Enfermedades raras, un enfoque práctico. Madrid: Instituto de Salud Carlos III, Instituto de Investigación de Enfermedades Raras; 2004. p. 13-5. [ Links ]
6. Burt RW. Polyposis syndromes. Clin Perspectives Gastroenterol 2002:51-9. [ Links ]
7. Starink, Van der Venn JP, Arwert F, de Waal LP, de Lange GG, Gille JJ, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet 1986;29:222-33. [ Links ]
8. Marra G, Armelao F, Vecchio FM, Percesepe A, Anti M. Cowdens disease with extensive gastrointestinal polyposis. J Clin Gastroenterol 1994;18:42-7. [ Links ]
9. Carlson GJ, Nivatvongs S, Snover Dc. Colorectal polyps in Cowdens disease (multiple hamartoma syndrome). Am J Surg Path 1984;8:763-70. [ Links ]
10. Mc Garrity TJ, Wagner Baker MJ, Ruggiero FM, Thiboutot DM, Hampel H, Zhou XP, et al. GI polyposis and glycogenic acanthosis of the esophagus associated with PTEN mutation positive Cowden syndrome in the absence of cutaneous manifestations. Am J Gastroenterol 2003; 98:1429-34. [ Links ]
11. Salem OS, Steck WD. Cowdens disease (multiple hamartoma and neoplasia syndrome): a case report and review of the English literature. J Am Acad Dermatol 1983; 8:686-96. [ Links ]
12. Botma M, Russel DI, Kell RA. Cowdens disease: a rare cause of oral papilomatosis. J Laringol Otol 2002; 116:221-3. [ Links ]
13. Hand JL, Rogers, RS. Oral manifestations of genodermatoses. Dermatol Clin 2003;21:183-94. [ Links ]
14. Thyresson HN, Doyle JA. Cowdens disease (multiple hamartoma syndrome) Mayo Clin Proc 1981;56:179-84. [ Links ]
15. Malone JP, Levin RJ, Fedok FG. Otolaryngologic manifestations of Cowden syndrome. Otolaryngol Head Neck Surg 2000;123:644-6. [ Links ]
16. Starink TM. Cowdens disease; analysis of fourteen new cases. J Am Acad Dermatol 1984;11:1127-41. [ Links ]
17. Lee HR, Moon YS, Yeom CH, Kim KW, Churi JY, Kim HK, et al. Cowdens disease. J Korean Med Surg 1997;12:570-5. [ Links ]
18. Attard TM, Lynch HT. Diagnosis and management issues in pediatric patients with gastrointestinal polyps. Pract Gastroenterol April 2003:57-72. [ Links ] 19. Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet 2000;37:828-30. [ Links ] 20. Schrager CA, Schneider D, Gruener AC, Tsou HC, Peacocke M. Clinical and pathological features of breast disease in Cowdens syndrome: An underrecognized syndrome with an increased risk of breast cancer. Hum Pathol 1998;29:47-53. [ Links ] 21. Marsh DJ, Coulon V, Lunetta KL, Roca-Serra P, Dahia PL, Zheng Z, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet 1998;7:507-15. [ Links ] 22. Hildenbrand C, Burgdorf WH, Lautenschlager. Cowden syndrome-Diagnostic skin signs. Dermatology 2001; 202:362-6. [ Links ] 23. Weary PE, Gorlin RJ, Gentry WC, Corner JE, Greer KE. Multiple hamartoma syndrome (Cowden disease). Arch Dermatol 1972;106:682-90. [ Links ] 24. Gorlin RJ, Cohen MM, Levin LS. Hamartoneoplastic syndromes: Cowden´s syndrome (multiple hamartoma syndrome). En: Syndromes of the Head and Neck. New York: Oxford University Press ed; 1990. p. 357-61. [ Links ] 25. Blanco V, Keochgerián V. Cowdens syndrome. Case report, with reference to an affected family. Med Oral Patol Oral Cir Bucal 2006;11:12-6. [ Links ] 26. Almenar R, Bagán JV, Milián MA, Jiménez Y. Síndrome de Cowden: presentación de un caso clínico con lesiones orales. An Med Int 2001;18:426-8. [ Links ] 27. Morrison JP, Kevin NC. Multiple endocrine neoplasia type IIB (mucosal neuroma syndrome, Wagenmann-Frobose syndrome). J Med Genet 1996;33:779-82. [ Links ] 28. Greer RO, Popper HA, de Mento FJ. Cowden´s disease (multiple hamartoma syndrome): report of a limited mucocutaneous form. J Periodontol 1976;47:531-4. [ Links ] 29. Hemmings CT. Thyroid pathology in four patients with Cowdens disease. Pathology 2003;35:311-4. [ Links ] 30. Eng C. Cowden syndrome. J Genet Counsel 1997;6:181. [ Links ] 31. Baù MG, Arisio R, Cristini G, Bertone E, Campogrande M. Screnning-detected breast carcinoma in a patient with Cowden síndrome. The breast 2004;13:239-41. [ Links ] 32. Saccardi A, Bacci S, Romagnoli P, Ravina A, Ficarra G. Cowden´s syndrome: a case report with clinical histopathological and immunological studies. Minerva Stomatol 1994;43:423-8. [ Links ] Recibido: 29-12-2005 ![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. Luis Miguel Capitán Cañadas
Servicio de Cirugía Oral y Maxilofacial
Hospital Universitario Virgen de las Nieves. CRT.
Carretera de Jaén s/n. Granada. España.
E-mail: luismxf@hotmail.com
Aceptado: 29-03-2006

