










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Internet)
versión On-line ISSN 1698-6946
Med. oral patol. oral cir.bucal (Internet) vol.12 no.2 mar. 2007
Langerhans cell histiocytosis in the maxillofacial area in adults. Report of three cases
José Antonio García de Marcos1, Alicia Dean Ferrer2, Francisco Alamillos Granados3, Juan José Ruiz Masera4, Gracia Barrios Sánchez5, Ana Isabel Romero Ortiz6, José María Calderón Bohórquez7, Borja Valenzuela Salas7
(1) Consultant. Department of Oral and Maxillofacial Surgery. Albacete University Hospital. Albacete
(2) Head of Section. Department of Oral and Maxillofacial Surgery. University Hospital "Reina Sofía". Cordoba. Fellow of the European Board of Oral and Maxillofacial Surgery. Associated Professor. School of Medicine. Córdoba University
(3) Consultant. Department of Oral and Maxillofacial Surgery. University Hospital "Reina Sofía". Cordoba, Spain. Fellow of the European Board of Oral and Maxillofacial Surgery. Associated Professor. School of Medicine. Córdoba University
(4) Consultant. Department of Oral and Maxillofacial Surgery. University Hospital "Reina Sofía". Cordoba
(5) Consultant. Department of Oral and Maxillofacial Surgery. La Ribera Hospital. Alcira. Valencia
(6) Resident. Department of Pathology. University Hospital "Reina Sofía". Cordoba
(7) Resident. Department of Oral and Maxillofacial Surgery. University Hospital "Reina Sofía". Cordoba. Spain
ABSTRACT
Langerhans cell histiocytosis (LCH) is a disease of unknown etiology, characterized by proliferation of pathological Langerhans cells within different organs. It mainly affects children, but adult cases also occur, with an incidence rate of one to two per million. The head and neck are affected in almost 90% of cases. Diagnosis is made by means of histopathological analysis, and imaging studies are necessary in order to determine extent of the disease. There are no controlled trials proposing an optimal treatment protocol for LCH. Prognosis in adults is generally good due to the slow evolution of the disease and its favourable response to treatment. In our report, we present three cases of LCH in patients aged 16, 24, and 28 years respectively, with primary manifestation in the maxillofacial area. A literature review was also conducted.
Key words: Langerhans cell histiocytosis, LCH, histiocytosis X, lymphoproliferative disease.
RESUMEN
La Histiocitosis de Células de Langerhans (HCL), es una enfermedad de etiología desconocida, que se caracteriza por la proliferación e infiltración anormal, de órganos, por células de Langerhans patológicas. Afecta predominantemente a pacientes en edad pediátrica, siendo en adultos la incidencia de la enfermedad de uno a dos casos por millón de habitantes. Las manifestaciones en cabeza y cuello aparecen en casi un 90% de los casos. El diagnóstico se obtiene por medios anatomopatológicos, siendo necesarias una serie de pruebas, determinantes de extensión, en todos los pacientes diagnosticados de HCL. No existen estudios, controlados, que determinen un tratamiento óptimo para la HCL. El pronóstico de esta enfermedad en adultos es generalmente bueno debido a la lenta evolución de la enfermedad y a su buena respuesta al tratamiento. Presentamos una revisión de tres casos de HCL, de 16, 24 y 28 años de edad, con manifestación primaria en el área Maxilofacial. Así mismo, realizamos una revisión de la literatura.
Palabras clave: Histiocitosis de células de Langerhans, LCH, Histiocitosis X, enfermedad linfoproliferativa.
Introduction
Langerhans cell histiocytosis (LCH) is a disease of unknown etiology, characterized by proliferation of pathological Langerhans cells within different organs (1-4). In 1953, Lichtenstein observed cytoplasmic bodies, known as X bodies, within histiocytes from tissues of patients suffering from what were previously considered distinct clinical disorders: eosinophilic granuloma, Hand-Schüller-Christian disease and Abt-Letterer-Siwe disease (2,3,5-8). As a result of their common underlying histopathology, Lichtenstein grouped these diseases together under the name of histiocytosis X. With Nezelof´s discovery in 1973 that these histiocytes were in fact Langerhans cells, the disorder was renamed Langerhans cell histiocytosis (2,7). LCH has also been referred to as Langerhans cell granulomatosis, histiocytosis X, Hashimoto-Pritzkers syndrome, non lipidic reticuloendotheliosis, type II histiocytosis, or self-healing histiocytosis (2,9).
LCH has a slight predilection for males and generally appears in childhood (1,4,6,8). The incidence of the disease in the adult population is from one to two cases per million inhabitants, ranging in presentation from 15 to 91 years, with mean age of diagnosis 35 years (4,10). Although lesions may appear in tissues of varying origins such as skin, hypothalamus, liver, lung, or lymphoid tissue (4,6,8,11,12), bone is the most common site of the disease (3,13). The head and neck are involved in almost 90 % of cases, and may be the only areas affected (2). The skull is involved in 50 % of cases; the temporal bone, meatal skin and cervical lymphatic nodes in 20-25 % of cases, while maxillary and mandibular bones are affected in 5 to 10 % of cases (2).
The Writing Group of the Histiocyte Society has identified three levels of confidence in the diagnosis of LCH, as follows: a presumptive diagnosis can be made when light morphologic characteristics are consistent with the findings defined in the literature for LCH; a designated diagnosis when, besides consistent light morphologic features, two or more supplemental positive stains for adenosinetriphosphatase, S-100 protein, alpha-D-mannosidase or peanut lectin are also present; and a definitive diagnosis when, besides light morphologic characteristics, Birbeck granules are detected in the lesional cell using electron microscopy and/or there is a positive staining for CD1a antigen (T6) on the surface of lesional cells (1,4). Once the condition has been diagnosed, adequate workup to determine the extent of the disease is mandatory (8,10). There are no controlled studies establishing optimal treatment for LCH (2,7,9,10). Prognosis of LCH in adults is generally good due to the slow evolution of the disease and its favourable response to treatment (11).
Clinical case reports. (table 1)
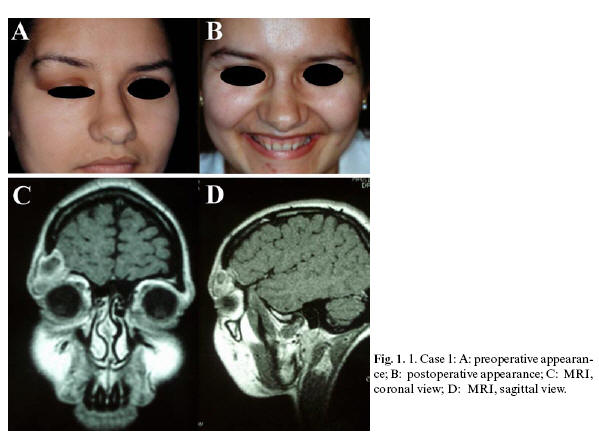
Case 1. A 24-year-old female was referred to the Dept. of Oral and Maxillofacial Surgery with a one-month history of swelling of the right upper eyelid, causing convex distortion of the eyebrow and adjacent frontal area. The lesion was elastic to the touch, painful, and impeded normal upper eyelid movement (Fig.1. A). Radiographic imaging showed a lobulated, lytic lesion in the right fronto-supraorbital region, with ground-glass appearance. There was no evidence of periosteal reaction or marginal sclerosis. Further examination with cranial MRI revealed the lesion to be 2.5 cm in diameter, circumscribed and cystic, with adjacent bone destruction in the right supraorbital area (Fig.1.C and D). Bone scintigraphy showed a single, hypercaptating lesion in the right supraorbital region. Biopsy revealed the diagnosis of LCH. After adequate workup to determine the extent of the disease, no other lesions were encountered. Resection was performed by superolateral orbitotomy, using a combined craniofacial approach. The resulting bony defect in the orbital roof and lateral orbital wall was grafted with calvarial bone and fixed by means of miniplates (Fig.1. B). Four years following surgery, the patient was diagnosed with hypogonadotropic hypogonadism and diabetes insipidus. Nine years post-surgery, there are no signs of recurrence of the lesion.
Case 2. A 16-year-old male was referred to the Dept. of Oral and Maxillofacial Surgery, affected by a sudden proptosis of the right eye with restriction of ocular movements, eyelid swelling and conjunctival hyperemia. CT scan showed a 3.5 cm mass in the superolateral region of the right orbit, making contact with the posterosuperior part of the right eyeball and right optic nerve, and spreading through the orbital roof into the anterior cranial fossa (Fig.2. A). A well-defined, hypercaptating, heterogeneous lesion was observed in the superolateral region of the right orbit on MRI (Fig.2. B). Biopsy revealed the diagnosis of LCH. Bone scintigraphy showed pathological focal hypercaptation in the area of the lesion, the latter resected by means of a combined craniofacial approach. A bicoronal flap was elevated and an orbito-zygomatic osteotomy exposed the tumor (Fig.2.C). After en bloc resection of the tumor, the orbital walls were reconstructed with outer-table calvarial bone grafts. Resorbable miniplates (Polimax®) were employed to fix the grafts and the bone flaps (Fig.2.D). One and a half years following surgery, there are no signs of relapse, nor complications associated with LCH.
Case 3. A 28-year-old male with previous history of right upper jaw odontogenic cyst removal at the site of dental pieces 15 and 16, with exodontia of same due to mobility. Three months following said intervention, the patient presented with pain and swelling in the treated area. An orthopantomogram showed extensive bone loss with respect to previous radiographs, involving right maxillary mesial surface of 17 to distal surface of 12 (Fig.3.A). The lesion was resected once again, and the histopathological report revealed LCH. Bone scintigraphy showed no other lesions. Four months following the second surgery, the patient presented with trismus, swelling and pain in the left upper jaw and in right and left mandibular bodies. On the orthopantomogram, new cystic lesions were observed in the regions of dental pieces 25, 36 to 37, and 46 to 48, with affectation of these teeth (Fig.3. B). Cystectomy of mandibular lesions was performed, as well as exodontia of dental pieces 36, 37, 46, 47 and 48 due to mobility.
The postoperative histopathological report revealed LCH. Subsequently, two additional surgeries were undertaken due to the appearance of new lesions in the right malar bone, right and left hemimaxillae, and left mandibular body (Fig.3. C). When yet more new lesions were discovered in the right mandibular angle, ramus and body (Fig.3. D), we decided to initiate chemotherapy treatment. The first phase of therapy consisted of intravenous vinblastine (6g/m2/week, with a total dose of 110g, in 11 cycles of 10 g each), with a second phase of oral methotrexate (2.5mg/3 times/week), which is currently being administered to the patient. A positive clinical and radiological response was observed soon after beginning chemotherapy 11 months ago, with no sign of recurrence of the lesions to date.
Discussion
The etiopathogenesis of LCH is unknown, although it appears to be linked to a disturbance in immune system regulation (2,3,8,12-14). In spite of being considered a disease of sporadic appearance, some authors have postulated the existence of a familiar predisposition (4,8,14).
Manifestation of LCH may take various forms. In the maxillofacial area, skin affectation may appear as a papular rash; scalp involvement has a seborrhoea-like presentation. Oral mucosa affectation, although infrequent, is characterized by gingival hypertrophy and ulcers of the buccal mucosa, hard and soft palates, and tongue (8,10,13). Osseous involvement is observed in 78% of patients, the cranium being the bone most commonly affected (49%). Bony lesions of the maxillomandibular area are also frequent, occurring in 30% of adult cases, particularly affecting posterior regions of the mandible (7,8,14). Pain and swelling of the mandible, with mobility and loss of teeth, may be the presenting symptoms of the disease (1,3,6,8,11,14). Intraorbital involvement can produce proptosis (3,8), as occurred in Case 2 of this report. The lymphatic system may be affected, with enlargement of lymphatic ganglions, Waldeyer´s ring or the thymus (2,8). Other soft tissues have been reported to be affected in the head and neck area, such as the eyelids, parotid and submandibular glands, the external auditory canal, the middle ear, the thyroid and the gastrointestinal tract (5,8,15).
Plain radiographs show LCH bony lesions as non-calcified, lytic areas without peripheral sclerosis. The radiological image of "floating teeth" is typical of maxillary and mandibular involvement (2,3,5,11,14,16). The differential diagnosis of mandibular lesions must include odontogenic cysts and tumors, primary bone tumors, osteomyelitis, metastases, multiple myeloma and giant cell granuloma (2,11,14). Radiographic imaging of lytic lesions of the skull reveals a punched-out pattern without evidence of periosteal reaction or marginal sclerosis, known as "geographic skull" (2,11). In some cases, a central mass of residual bone can be seen within the lytic lesion, giving the appearance of a "bull´s eye", alternatively referred to as a "button sequestrum" or a "hole within a hole" (3). The soft tissue mass accompanying all osseous lesions can best be seen with CT scan and MRI (3), the latter showing a well-defined area of soft tissue surrounding a focal lesion, with altered bone marrow signal (2). Scintigraphy is useful for evaluating extent of the disease and for monitoring its evolution (7).
Histological features of LCH lesions include, besides Langerhans cells, the presence of a variable number of eosinophils, neutrophils, mononuclear and polynuclear histiocytes, and lymphocytes (1,3,6,8,11). Initial stage lesions tend to be very cellular, whereas in more advanced stages, a higher degree of fibrosis is present (6). Histological appearance does not correlate with clinical behaviour (11). Langerhans cells are oval or rounded in shape, pale, and have a predominantly eosinophilic cytoplasm (2,8). The nucleus is oval or lobulated, with a typical central sulcus, giving a "coffee bean" appearance. Birbecks granules, which can be seen inside Langerhans cells with electron microscopy, are pathognomonic for LCH (8,11). These are trilamellar, cytoplasmatic structures with either an elongated or tennis racket shape (if there is dilation of one end), characterized by periodic zipper-like striations (11). Langerhans cells also express S-100 protein, ATP-ase, peanut lecithin, alpha-D-mannosidase, and antigens CD 207 and CD1a (2,3,8,14).
Multiple therapeutic modalities have been suggested for the treatment of LCH, such as curettage, resection, radiotherapy, chemotherapy, and intralesional and systemic corticosteroids (8,11,12). For patients with monostotic disease, some authors recommend therapeutic abstention, since spontaneous regression takes place in the majority of these cases. Others advocate surgical treatment of the lesion by means of curettage or biopsy (4,6,7,11,12,14), or intralesional injection of corticosteroids (2,6,11,12,14). Radiotherapy, in doses of 1,200 to 1,800 cGys, has been proposed for non-accessible lesions, for those which compromise vital structures such as the optic nerve, and for recurrence of previously resected lesions (6,9,11). When lymphatic nodes are involved, the most adequate treatment is excision of affected nodes. In cases of skin manifestation only, topical steroids and intralesional interferon-beta can be employed (6,17), although success with oral thalidomide has also been obtained in cutaneous LCH (8). Oral mucosal lesions without underlying bone disease respond well to perilesional infiltration of triamcinolone acetonide (13).
For patients with multiple LCH lesions, there is no universally accepted single treatment strategy. Some clinicians advocate aggressive, generalized treatment in these cases, whereas others suggest a conservative, symptomatic approach, unless there are systemic manifestations such as pain, fever, failure to thrive or disorders of vital organs. While radiation alone, or single-drug administration have been shown to be insufficient for treatment of multiple bony lesions, most of these patients respond to therapy with a combination of steroids and cytotoxic agents (2,18). For example, the rate of recurrence of the disease falls markedly with 6-month treatments of prednisone and vinblastine. Other studies defend the effectiveness of 2-chloro-2´-deoxyadenosine (2CdA) in patients with recurrent and multisystemic affectation (8,10).
In terms of prognosis, both unifocal and multifocal lesions respond well to treatment, with cure rates of up to 80%. However, 20% of patients develop a more aggressive, multiorganic affectation which requires repeated treatments, and produces a 9% mortality rate (8,11). In the majority of refractory or life-threatening cases, there is lung involvement (11), with a worse prognosis for isolated lung affectation than for multisystemic disease, even if there are lung lesions in the latter case (4). In contrast, cases with isolated bone involvement have cure rates of 97% (11). The overall 5-year survival rate for LCH is approximately 92.3% (4).
In all three cases presented in this report, the lesions were surgically resected. In Cases 1 and 2, there has been no evidence of local relapse in 9 years and 1 year of disease evolution, respectively. However, in Case 3, due to the rapid and multiple bony relapses in the maxillofacial area following surgeries, we administered chemotherapy consisting of vinblastine and methotrexate, with favourable results.
Given the rarity of LCH, we strongly recommend protocolization of treatment. To further this end, patients suffering from this disease, particularly those with multiorganic affectation, should be included in clinical trials carried out by the Histiocytosis Society.
References
1. Cotton RT, Rothschild MA, Zwerdling T, Ballard ET, Myer CM, Koch BL.Tumors of the Head and Neck in children. En: Thawley SE, Panje WR, Batsakis JG, Lindberg RD. Comprehensive management of Head and Neck Tumors. Philadelphia: Second edition. W.B. Saunders Company; 1999. p. 1865-8. [ Links ]
2. O´Hare TJ. Granulomatous and lymphoproliferative diseases of the Head and Neck. En: Thawley SE, Panje WR, Batsakis JG, Lindberg RD. Comprehensive management of Head and Neck Tumors. Philadelphia: Second edition. W.B. Saunders Company; 1999. p. 1966-8.
3. Devaney KO, Putzy MJ, Ferlito A, Rinaldo A. Head and Neck Langerhans Cell Histiocytosis. Ann Otol Laryngol 1997;106:526-32.
4. Aricò M, Girschikofsky M, Généreau T, Klersy C, McClain K, Grois N, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 2003;39: 2341-8.
5. Miller ML, Sassani JW, Sexton FM. Diffuse Histiocytosis X involving the eyelid of 65-year-old Woman. Am J Ophthalmol 1992;15;113:458-9.
6. Key SJ, O´Brien CJ, Silvester KC, Crean S-J. Eosinophilic granuloma: resolution of maxillofacial bony lesions following minimal intervention Report of three cases and a review of the literature. J CranioMaxillofac Surg 2004;32:170-5.
7. Bartnick A, Friedrich RE, Roeser K, Schmelzle R. Oral Langerhans cell histiocytosis. J CranioMaxillofac Surg 2002;30:91-6.
8. Hicks J, Flaitz CM. Langerhans cell histiocytosis: Current insights in a molecular age with emphasis on clinical oral and maxillofacial pathology practice. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2005;100:S42-66.
9. Seegenschmiedt HM, Micke O, Olschewski T, Bruns F, Heyd R, Schaefer U, et al. Radiotherapy is effective in symptomatic Langerhans Cell Histiocytosis (LCH): Long term results of a multicenter study in 63 patients. Int J Radiat Oncol Biol Phys 2003;57,2;1: S251.
10.McClain K. Adult Langerhans Cell Histiocytosis. Histiocyte Society. 1999-2005. "www.histio.org/society/LCH/Adult/mcclain2.shtml".
11. Neoplasm of the immune system. En: R.E. Marx, D. Stern. Oral and Maxillofacial Pathology. A rationale for diagnosis and treatment. Illinois:Quintessence Publishing Co, Inc; 2003. p. 870-5.
12. Harris GJ, Woo KI. Eosinophilic granuloma of the orbit: a paradox of aggressive destruction responsive to minimal intervention. Trans Am Ophthalmol Soc 2003;101:93-106.
13. Milián MA, Bagán JV, Jiménez Y, Pérez A, Scully C, Antoniades D. Langerhans cell histiocytosis restricted to the oral mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001;91:76-9.
14. Eckardt A, Schultze A. Maxillofacial manifestations of Langerhans cell histiocytosis: a clinical and therapeutic analysis of 10 patients. Oral Oncol 2003;39:687-94.
15. Green I, Behar AJ, Shanon E, Gorsky M. Multifocal Extraosseous Eosinophilic Granuloma of the Head and Neck. Arch Otolaryngol Head Neck Surg 1988;114:561-3.
16. Hernández-Juyol M, Boj-Quesada JR, Gallego-Melcon S. Manifestaciones orales de la Histiocitosis de células de Langerhans. A propósito del caso de un niño de dos años. Med Oral 2003;8:19-25.
17. Matsushima Y, Baba T. Resolution of cutaneous lesions of histiocytosis X by intralesion injections of interferon-beta. Int J Dermatol 1991;30:373-4.
18. Green JD, Neel III HB, Witzig TE. Lymphoproliferative Disorders of the Head and Neck. Am J Otolaryngol 1991;12:26-32.
![]() Correspondence:
Correspondence:
Dr. Jose A. García de Marcos
C/ Antonio Acuña. Nº10.5ºAizq.
28009. Madrid, España.
E-mail: pepio2@hotmail.com
Received: 24-12-2005
Accepted: 5-01-2007

