










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Clínica de Medicina de Familia
versión On-line ISSN 2386-8201versión impresa ISSN 1699-695X
Rev Clin Med Fam vol.4 no.2 Barcelona jun. 2011
Trisomia X, asociada a Dismorfismo Fenotípico
Trisomy X, related to dysmorphic phenotype
María Isabel Buedo Rubioa, Josefa Plaza Almeidab, Ma Carmen Carrascosa Romeroc, Blanca Alfaro Ponced
aResidente de Pediatría. Centro de Salud Zona 8 de Albacete (Albacete).
bPediatra. Centro de Salud Zona 8 de Albacete (Albacete).
cNeuropediatra Neonatal. Complejo Hospitalario Universitario de Albacete (Albacete).
dNeonatología. Complejo Hospitalario Universitario de Albacete (Albacete).
Dirección para correspondencia
RESUMEN
El síndrome triple XXX es una anomalía cromosómica relativamente frecuente, con una incidencia de 1 por cada 1.000 ó 1.200 recién nacidas vivas, en relación generalmente a edad materna elevada. Sin embargo, no suele sospecharse al nacimiento al no presentar un fenotipo característico y, aunque el dismorfismo puede ser muy variable, lo habitual es que no presenten ninguna manifestación clínica. Es por esto que el diagnóstico, con frecuencia, se establece tardíamente tras la presentación de insuficiencia ovárica primaria. Sólo en pocas ocasiones se describe en la literatura dismorfismo facial y otras malformaciones asociadas, sobre todo a nivel de aparato genitourinario, como genitales ambiguos, disgenesia ovárica, extrofia de cloaca, agenesia renal y con menor frecuencia cardiopatías u otras. El pronóstico es variable, dependiendo de la severidad de las anomalías presentadas, aunque hay casos descritos con retraso mental. Lo más frecuente es que tengan una inteligencia normal o ligeramente inferior a la normal.
Presentamos el caso de una niña diagnosticada en periodo neonatal por un síndrome polimalformativo, en la cual se han encontrado anomalías poco habituales en este síndrome, como un dismorfismo facial peculiar, la implantación anómala del pulgar, la hipoacusia bilateral y una cardiopatía congénita. También presenta, como hallazgos más frecuentes en este síndrome, retraso psicomotor y del lenguaje. Destacamos la importancia del diagnóstico precoz para la instauración temprana del tratamiento.
Palabras clave: Trisomía X, Cromosomopatías.
ABSTRACT
The triple X syndrome is a relatively common chromosome abnormality, with an incident of 1 per 1,000 or 1,200 live new born infants, generally related to advanced maternal age. However, it is not usually suspected at birth due to the lack of a characteristic phenotype and, although dysmorphism may be variable, there is usually no clinical manifestation. This is why diagnosis is often delayed until after the presentation of primary ovary failure. Facial dysmorphism and other related malformations, especially of genitourinary system, such as ambiguous genitalia, ovarian dysgenesis, cloacal exstrophy, renal agenesis and less frequently heart diseases or other diseases, have been rarely reported in the literature. The prognosis is variable, depending on the severity of the abnormalities, although cases of mental retardation have been described. Most patients have normal or slightly below normal intelligence.
We report a case of a girl diagnosed in the neonatal period with polymalformation syndrome, which included rare abnormalities, such as a peculiar facial dysmorphism, abnormal implantation of the thumb, bilateral hearing loss and congenital heart disease. The more common findings in this syndrome were also present such as psychomotor and language retardation. We emphasise the importance of early diagnosis for prompt installation of treatment.
Key words: Trisomy X, Chromosomal Abnormalities.
Introducción
El Síndrome 47 XXX fue descrito por primera vez en 1959 por Jacobs en una mujer con inteligencia normal y amenorrea1. Su prevalencia se ha estimado aproximadamente en 1 de cada 1.000 nacidas vivas1-5, considerando que tan sólo el 10% de los individuos con trisomía X se diagnostica actualmente2.
Esta cromosomopatía surge de la no disyunción del cromosoma X durante la división celular en el momento de la gametogénesis o tras la concepción (conocida como no disyunción post-cigótica). Estudios realizados al respecto han demostrado que el origen de este cromosoma adicional deriva en el 58-63% de los casos de un error en la meiosis I materna, el 16-17,4% en la meiosis II y el 18-19,6% son debidas a un error en la separación post-cigótica. Este tipo de errores se incrementa con la edad materna, siendo éste el principal factor de riesgo3.
Observaciones clínicas
Se presenta el caso de una niña, hija de madre de 28 años, secundigesta, con embarazo en riesgo teratogénico debido al tratamiento con ácido valproico durante la gestación por enfermedad epiléptica conocida. Diagnosticada, además, de síndrome ansioso-depresivo y trastorno fóbico adaptativo y de afectividad, recibiendo tratamiento prolongado con diacepam y alprazolam. Padre positivo para VHC.
Nacida mediante cesárea programada por presentación anómala a las 37+1 semanas de edad gestacional, tras un embarazo controlado que cursó sin incidencias. El peso al nacer fue de 2610 g (p25-50), la talla de 48 cm (p50) y el perímetro cefálico de 33 cm (p50).
Se requirió reanimación de tipo III al nacimiento por depresión respiratoria, colocándose CPAP tras su ingreso en UCIN. Las exploraciones complementarias fueron compatibles con enfermedad de membrana hialina y maladaptación pulmonar. Presentó ictericia no inmune al tercer día de vida, tratada con fototerapia. Durante su ingreso se observó un refuerzo del 2o tono cardíaco, por lo que se realizó ecocardiograma en el que se objetivó la presencia de foramen oval permeable/comunicación interauricular ostium secundum y ductus arterioso persistente, sin repercusión hemodinámica.
A la exploración física se apreció un fenotipo peculiar con base nasal ancha y aumento de la distancia intercantal, con hipertelorismo, micrognatia, cuello corto, implantación baja del cuero cabelludo y pulgares de implantación anómala, por lo que se decidió solicitar un estudio genético, cuyo cariotipo presentaba 47 cromosomas con fórmula sexual XXX. Además, se completó el estudio en busca de malformaciones con una ecografía cerebral, una ecografía abdominal, un electroencefalograma, un hemograma, una bioquímica con perfil hepático y renal y un estudio de coagulación, con resultados normales.
Las otoemisiones acústicas resultaron patológicas, por lo que se realizaron potenciales evocados auditivos, hallándose una hipoacusia de transmisión con afectación bilateral, más acusada en el oído izquierdo, con una pérdida auditiva del 98%.
A los 9 meses de vida se valoró en la consulta de Neurología Neonatal, objetivándose un ligero retraso global del desarrollo, con hipotonía. En este momento se remitió a los Servicios de Bienestar Social, aconsejándose inicio de estimulación y fisioterapia.
A los 2 años se diagnosticó de exotropia alternante y miopía, tras ser estudiada por el servicio de Oftalmología.
Actualmente se encuentra en seguimiento por las consultas de Cardiología, Neuro-Neonatología, Oftalmología y Otorrinolaringología, con terapia instaurada a cargo del Centro de Desarrollo infantil y Atención temprana, donde recibe fisioterapia, logopedia, estimulación, orientación y apoyo familiar.
Comentarios
Las mujeres con cariotipo 47 XXX tienen una alteración del número de cromosomas sexuales que consiste en la presencia de un cromosoma X supernumerario2. No se encuentran rasgos dismórficos característicos, sin embargo, una minoría de los pacientes puede presentar, como en nuestro caso, rasgos dismórficos variables que no conforman un fenotipo específico (tabla 1),los más habitualesa nivel de aparato genitourinario, como genitales ambiguos, disgenesia ovárica, extrofia de cloaca, agenesia renal y, con menor frecuencia, cardiopatías, etc6. En nuestra paciente destaca el hipertelorismo, la baja implantación del cabello, cuello corto e hipotonía, entre otras malformaciones. Además, presenta una implantación anómala del pulgar, dato que no hemos encontrado descrito en la literatura (figura 1).

Tabla 1. Rasgos asociados a la Trisomía X. Fuente: Tartaglia NR,
Howell S, Sutherland A, Wilson R, Wilson L. A review of trisomy X (47 XXX).
Orphanet Journal of Rare Diseases. 2010; 5:8.
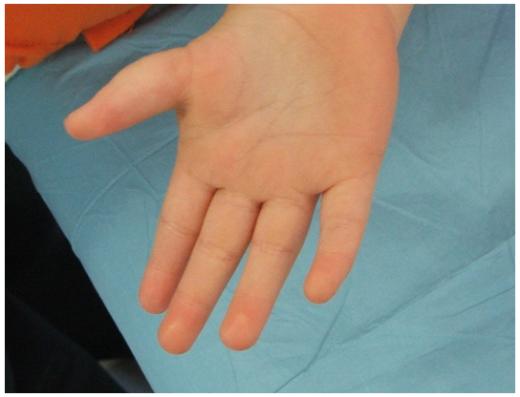
Figura 1. Implantación anómala del pulgar.
Estas niñas suelen tener una talla y un peso acorde con su edad, pero es típico el hallazgo de percentiles superiores al 75 en estatura, hecho que se hace más evidente en la adolescencia3. El desarrollo sexual y la fertilidad suelen ser normales, si no hay malformaciones de genitales internos, pero muestran un mayor riesgo de tener un fallo ovárico prematuro2. Es en este momento en el que se suele llegar al diagnóstico, por presentar una menarquia tardía.
El retraso mental es raro, aunque el cociente intelectual suele ser de 10-15 puntos por debajo de lo normal, y aproximadamente en la mitad de los casos se encuentran problemas de aprendizaje, con dificultad en el lenguaje y en el área motora2-3. Nuestra niña presenta un retraso global del desarrollo, más significativo en el área del lenguaje, sin anomalías del comportamiento.
En la mayoría de los casos no se aprecian anomalías importantes, no obstante, lo más frecuente es encontrar displasias renales y malformaciones ováricas, siendo menos comunes las malformaciones cardíacas. Dentro de estas últimas, hay que destacar los defectos del septo auricular, la estenosis pulmonar y la coartación de aorta3. A pesar de la baja frecuencia de cardiopatías congénitas, se describen en la literatura series de casos en los que, como en el nuestro, éstas pueden ser el único hallazgo malformativo importante en la clínica1.
La causa de la variabilidad fenotípica no está clara, aunque se relaciona con la inactivación del cromosoma X femenino. En todas las mujeres siempre se inactiva por azar uno de los dos cromosomas X, hecho que lleva a la inhibición de uno de los cromosomas X, lo cual es necesario para un desarrollo normal. En las niñas con trisomía X se inactivan dos de los tres cromosomas y se produce un mosaicismo en los tejidos, y esto podría ser una de las posibles explicaciones de los diferentes hallazgos4. Aunque el cariotipo 47 XXX sin mosaicismo es el más frecuente, éste ocurre aproximadamente en el 10% de los casos y puede aparecer en varias combinaciones, tales como 46 XX/47 XXX o 47 XXX/48XXXX, o en aquellas que incluyen líneas celulares con Síndrome de Turner, con 45 X/47XXX o 45 X/46XX/47XXX3.
El diagnóstico viene de la mano del estudio genético, con la realización del cariotipo tras la sospecha clínica, como en nuestra paciente. Sin embargo, se puede realizar ya intraútero, mediante la amniocentesis o la biopsia de vellosidades coriónicas3,7. Por otro lado, las anormalidades ecográficas que permiten el diagnóstico son extremadamente raras, siendo el diagnóstico genético un hallazgo casual en la mayoría de los casos7. Es importante identificar el mosaicismo con el Síndrome de Turner (45 X), con el fin de proporcionar la evaluación y el tratamiento que este síndrome requiere3.
La evolución de las niñas con cariotipo 47 XXX es favorable en la mayoría de los casos, sobre todo si no hay malformaciones concomitantes. Se ha observado que en una situación familiar adecuada se minimizan los problemas asociados2. En nuestro caso ha sido importante la estabilización del ambiente familiar y social de la niña, intentando en todo momento no tratar a la paciente de forma diferente, para facilitar la adaptación familiar.
La detección temprana de los posibles problemas de aprendizaje de estas niñas es importante para poder instaurar la terapia adecuada en cada caso, cuidando el desarrollo intelectual y tratando de asegurar el correcto desarrollo psicomotor. Es en este punto en el que el pediatra del centro de salud debe estar presente, facilitando el acceso a los Servicios de Bienestar Social si esto fuera necesario.
Bibliografía
1. Goldschmidt E, Márquez M, Solari A, Ziembar MI, Laudicina A. Variabilidad fenotípica en pacientes 47, XXX. Presentación de cuatro casos nuevos. Arch Argent Pediatr. 2010; 108(4):e88-e91. [ Links ]
2. Martínez-Frías ML, Rodríguez L, López F. Diagnóstico citogenético con resultado 47, XXX. Información general sobre las personas con esta alteración de los cromosomas sexuales. Madrid: Ed. Estudio Colaborativo Español de Malformaciones Congénitas; 2000. [ Links ]
3. Tartaglia NR, Howell S, Sutherland A, Wilson R, Wilson L. A review of trisomy X (47 XXX). Orphanet Journal of Rare Diseases. 2010; 5:8. [ Links ]
4. Jorde LB, Carey JC, Bamshad MJ, White RL. Genética Médica. 3a Ed. Barcelona: Elsevier; 2005. p. 88-90. [ Links ]
5. Jones KL. SMITH Patrones reconocibles de malformaciones humanas. Sexta Edición. Madrid: Elsevier Saunders; 2007. p. 72-3. [ Links ]
6. Lin HJ, Ndiforchu F, Patell S. Exstrophy of the cloaca in a 47, XXX child: review of genitourinary malformations in triple-X patients. Am J Med Genet. 1993; 45(6):761-3. [ Links ]
7. Ben Hamouda H, Mkacher N, Elghezal H, Bannour H, Kamoun M, Soua H et al. Prenatal diagnosis and prognosis of triple X syndrome: 47, XXX. J Gynecol Obstet Biol Reprod (Paris). 2009; 38(7):599-603. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
María Isabel Buedo Rubio,
Josefa Plaza Almeida
Centro de Salud Zona 8,
C/ Graduados s/n,
02006- Albacete, España.
Telf.: 967502745
e-mail: mib22n@hotmail.com
Recibido el 13 de enero de 2011.
Aceptado para su publicación el 10 de marzo de 2011.

