










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Clínica de Medicina de Familia
On-line version ISSN 2386-8201Print version ISSN 1699-695X
Rev Clin Med Fam vol.9 n.2 Barcelona Jun. 2016
UN PACIENTE CON...
Neuromielitis óptica de Devic
Devic neuromyelitis optica
Ruth Bravo-Lizcanoa, Lucía Sierra-Santosb, Lorraine Gil-Guliasa y Antonio L. Aguilar-Sheac
aM.I.R. Medicina Familiar y Comunitaria. Centro de Salud Reina Victoria. Madrid (España).
bMédico de Familia. Centro de Salud Manzanares el Real. Madrid (España).
cMédico de Familia. Centro de Salud Puerta de Madrid. Alcalá de Henares. Madrid (España).
Dirección para correspondencia
RESUMEN
La neuromieltis óptica (NMO) o enfermedad de Devic es un trastorno autoinmune, inflamatorio y desmielinizante del sistema nervioso central, que afecta principalmente al nervio óptico y la médula espinal. Considerada anteriormente como una variante de esclerosis múltiple (EM), hallazgos recientes han ayudado a concluir que la NMO es una entidad que presenta importantes diferencias inmunopatológicas, clínicas, de pronóstico y de respuesta al tratamiento, en comparación con la EM. El reciente descubrimiento y medición del anticuerpo antiacuaporina 4 (anti-AQP4 o también, NMO-IgG) ha reavivado el interés científico y activado la investigación de nuevas líneas de tratamiento de la NMO.
Presentamos el caso clínico de un paciente varón de 10 años que acude a la consulta por presentar, tras una caída accidental, gonalgia bilateral, dorsalgia y lumbalgia que impresionan inicialmente de contusiones y que, sin embargo, tras 48 horas evolucionan negativamente con persistencia de dolor, pérdida de fuerza y parestesias en los miembros inferiores, dificultad para incorporarse, incapacidad para deambular e incontinencia urinaria. Ante la sospecha de mielitis transversa se deriva al paciente al servicio de urgencias hospitalario, donde desarrolla a las 24 horas de ingreso una neuritis óptica derecha; finalmente es diagnosticado de neuromielitis óptica o enfermedad de Devic.
Palabras clave: Enfermedades desmielinizantes. Enfermedad de Devic. Mielitis. Neuritis óptica. Neuromielitis óptica.
ABSTRACT
Neuromyelitis optica (NMO) or Devic's disease is an autoimmune, inflammatory and demyelinating disorder of the central nervous system that affects mainly the optic nerve and the spinal cord. NMO was first considered a form of multiple sclerosis (MS). However, recent findings have led to the conclusion that NMO is a distinct disorder, presenting important immunopathological, clinical, prognostic and therapeutic differences from MS. The recent discovery and measurement of the anti-aquaporine-4 antibody (anti-AQP4 or NMO-IgG) has revived the scientific interest and has boosted research of new lines of treatment for NMO. We present the clinical case of a ten year-old boy who, after an accidental fall, presents bilateral knee pain, and pain in the upper and lower back, which at first is thought to be a contusion. However, after 48 hours it develops negatively, persisting the pain, loss of strength and lower limb paraesthesia, difficulty in sitting up, inability to walk, and urinary incontinence. Transverse myelitis is suspected and the patient is sent to the hospital emergency service, where after 24 hours he develops a right optic neuritis. Finally, a neuromyelitis optica or Devic's disease is diagnosed.
Key words: Demyelinating diseases. Devic Disease. Myelitis. Optic neuritis. Neuromyelitis optica.
Introducción
La neuromielitis óptica (NMO) de Devic es un trastorno autoinmune inflamatorio desmielinizante, anteriormente considerado como una forma de esclerosis múltiple (EM). Actualmente se sabe que presenta importantes diferencias con la EM y, aunque el diagnóstico diferencial pudiera ser en ocasiones difícil, la evolución clínica y la medición del anticuerpo antiacuaporina 4 (anti-AQP4) son claves para su diagnóstico1-4.
Caso clínico
Presentamos el caso de un varón de 10 años sin antecedentes de interés, con gonalgia bilateral, dorsalgia y lumbalgia tras caída de una bicicleta hace 4 días. A la exploración presentaba una movilidad activa y pasiva conservada con dolor en ambos miembros inferiores (MMII). No se observaron datos de fracturas ni alteraciones neurológicas. Se pautó reposo deportivo, frío local y tratamiento antiinflamatorio. A las 48 horas, el paciente acude nuevamente por persistencia del dolor, pérdida de fuerza, parestesias en MMII, dificultad para incorporarse y deambular e incontinencia urinaria. La exploración física reveló una disminución de la fuerza y una alteración de la sensibilidad en ambos MMII. Ante la sospecha de mielitis transversa se derivó al paciente al servicio de urgencias hospitalario. Durante su ingreso la analítica sanguínea resultó normal, el estudio del líquido cefalorraquídeo reveló una moderada pleocitosis, sin presencia de bandas oligoclonales de IgG; el electroencefalograma no evidenció alteraciones y los potenciales evocados demostraron una parálisis y espasticidad de MMII, hiporreflexia y Babinsky bilateral, ausencia de reflejos abdominales y cremastéricos, hiperalgesia y disestesia del tronco y MMII. El electromiograma reveló una alteración de la sensación somatosensorial medular a través de los cordones posteriores, sin conducción a través del cordón derecho y con alteración parcial del cordón izquierdo. La resonancia magnética (RM) cerebral resultó normal, sin embargo, la RM medular mostró una lesión expansiva a nivel medular desde D1 a D6 compatible con mielitis transversa (figura 1).
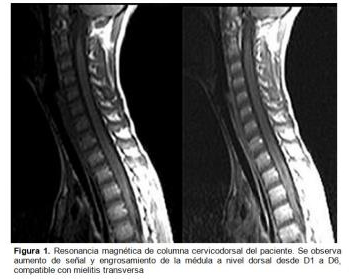
A las 24 horas de ingreso, el paciente presentó un dolor intenso en el ojo derecho con pérdida brusca de la agudeza visual. Fue valorado por oftalmología, diagnosticándose una neuritis óptica derecha.
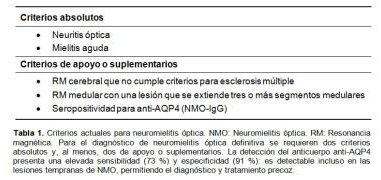
Ante la presencia de mielitis transversa y neuritis óptica, con RM cerebral que no cumplía criterios de esclerosis múltiple y RM medular que muestra una lesión de tres o más segmentos medulares (D1 a D6), se realizó estudio del anticuerpo antiacuoporina-4, que fue negativo. De acuerdo con la clínica, evolución y pese a la negatividad del anticuerpo, se diagnosticó al paciente de neuromielitis óptica o enfermedad de Devic (ver criterios diagnósticos tabla 1).
Inicialmente el paciente fue tratado con metilprednisolona intravenosa a dosis de 1 g/día sin presentar mejoría, por lo que se realizó plasmaféresis con buena evolución clínica.
El paciente fue seguido por neurología, oftalmología y urología con mejoría progresiva y descenso progresivo de corticoterapia. Catorce meses después, el paciente presentaba una recuperación completa de fuerza y sensibilidad de MMII, marcha normal, reflejos normales, ausencia de espasticidad, ataxia o dismetría; ocasionalmente presentaba hiperestesia lumbar dolorosa de carácter autolimitado. En el ojo derecho mantiene visión borrosa de cerca, con atenuación de la percepción de los colores, aunque consiguió recuperar el 70 % de la agudeza visual. Persiste la enuresis nocturna por espasticidad vesical que continúa tratando con desmopresina.
Discusión
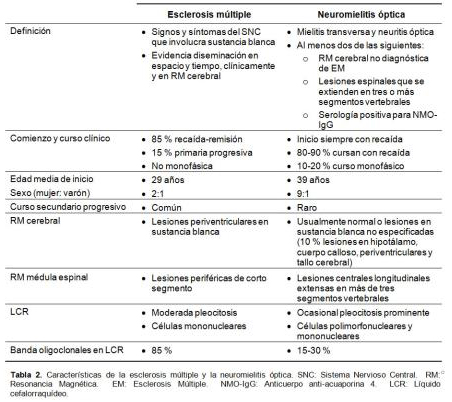
La NMO o Enfermedad de Devic fue descrita en 1984 por el médico francés Eugéne Devic2. Es una enfermedad inflamatoria, autoinmune y desmielinizante del sistema nervioso central (SNC) que se caracteriza por ataques de neuritis óptica (NO) y mielitis3-5. Aunque inicialmente se consideró que podría corresponder a una variante de esclerosis múltiple, actualmente se acepta que es una entidad totalmente diferente (tabla 2)2-5. A esta diferenciación ha contribuido el descubrimiento en 2004 de los anticuerpos antiacuaporina-4 (anti-AQP4 o NMO-IgG), muy sensibles y específicos, presentes en el 70 % de los pacientes con NMO, y que se encuentran en una minoría de quienes padecen EM1-4. La diferencia fisiopatológica de la NMO es que el ataque no es mediado por células T, sino por células B2-4.
La NMO tiene una distribución mundial y una prevalencia estimada de 1-2/100.000 habitantes. Es más común en individuos no caucásicos3, y se asocia a diversas enfermedades autoinmunes e infecciones vírales2-3.
La NMO puede presentarse en forma monofásica (30 %) o en brotes (70 %)1-5. En la forma monofásica los pacientes experimentan, prácticamente de forma simultánea, una neuritis óptica unilateral o bilateral y un episodio de mielitis transversa, sin que sea habitual la presencia de recaídas posteriores; esta forma de presentación es más frecuente en pacientes jóvenes, como en nuestro caso. Sin embargo, en los pacientes con un curso en brotes, lo más común es que la presentación de ambos episodios (neuritis óptica y mielitis transversa) sea secuencial y no simultánea, con un intervalo de presentación entre ambos eventos de años o décadas, siendo más frecuente en mujeres de mediana edad1.
El cuadro clínico típico de NMO es la presencia de dolor ocular, más frecuentemente unilateral, con pérdida de agudeza visual y que precede al episodio de mielitis con cuadriparesia o paraparesia, pérdida de la sensibilidad por debajo de la lesión y disfunción vesical. En nuestro paciente, primero se presentó la afectación medular y posteriormente la neuritis óptica.
El diagnóstico se basa en hallazgos clínicos y de imagen, propuestos en 1999 por la Clínica Mayo y que fueron actualizados en 2006 tras el descubrimiento de los anticuerpos anti-AQP4 (tabla 1), con una sensibilidad del 99 % y una especificidad del 90 %1-6.
La NMO continúa siendo una enfermedad incurable. El objetivo del tratamiento es el control de los síntomas agudos, la prevención médica de las complicaciones y la rehabilitación4.
El tratamiento de la fase aguda puede iniciarse con esteroides intravenosos (metilprednisolona 1 g/día 3-5 días), seguido de pauta descendente de prednisolona oral2. La plasmaféresis intravenosa debe considerarse en pacientes con mala respuesta al tratamiento inicial con corticoesteroides, o en aquellos que presentan un importante deterioro con pérdida visual aguda grave o mielitis cervical severa con riesgo de depresión respiratoria de origen neurogénico1.
Los pacientes con buena respuesta al tratamiento con plasmaféresis, pueden ser tratados en brotes sucesivos con dicho tratamiento como primera opción4.
Para prevenir nuevos episodios de NMO los tratamientos se basan en inmunosupresores como azatrioprina o rituximab4, cuyo uso se recomienda antes incluso del diagnóstico definitivo de NMO tras haber presentado un primer ataque de neuritis óptica o mielitis aisladas, en pacientes con seropositividad para anti-AQP41-2.
En muchos casos, el pronóstico de la enfermedad es nefasto. Las recaídas ocurren frecuentemente dentro de los primeros tres a cinco años tras el inicio de la enfermedad; más del 30 % de los pacientes no diagnosticados o no tratados pueden morir por un ataque de mielitis severa con insuficiencia respiratoria (principal causa de muerte de la NMO) y más del 50 % de los pacientes quedan ciegos de uno o ambos ojos, a causa de la NMO1.
Como conclusión queremos resaltar que la NMO es una entidad diferente a la EM cuyo diagnóstico y tratamiento precoz, así como el progresivo avance de la investigación sobre la fisiopatología de la misma, es vital para lograr un adecuado conocimiento, manejo y tratamiento de esta aún desconocida y grave enfermedad. Desde atención primaria debemos conocer y descartar una posible NMO ante cualquier paciente que presente un episodio de neuritis óptica o mielitis transversa, ya sea de forma aislada o simultánea, derivando rápidamente al servicio de urgencias del hospital de referencia, para estudio, diagnóstico e inicio precoz del tratamiento que evite sus graves complicaciones.
Bibliografía
1. Chiquete E, Navarro-Bonnet J, Ayala-Armas R, Gutiérrez-Gutiérrez N, Solórzano-Meléndez A, Rodríguez-Tapia D, et al. Neuromielitis óptica: actualización clínica. Rev Neurol, 2010; 51 (5): 289-94. [ Links ]
2. Pinzón A, Echeverry T, Rodriguez AB. Neuromielitis óptica (enfermedad de Devic). Acta Med Colomb. 2010; 35 (1): 21-5. [ Links ]
3. Lopategui Cabezas I, Cervantes Llano M, Pentón Rol G. Neuromielitis óptica. Principales diferencias con la esclerosis múltiple. An Med Interna (Madrid). 2008; 25 (6): 294-6. [ Links ]
4. Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. 2014; 176 (2): 149-164. [ Links ]
5. Etemadifar M, Nasr Z, Khalili B, Taherioun M, Vosoughi R. Epidemiology of neuromyelitis optica in the world: a systematic review and meta-analysis. Mult Scler Int. 2015: 174720. [ Links ]
6. Wingerchuck DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006; 66 (10): 1485-9. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ruth Bravo Lizcano.
Correo electrónico: ruth.b.l.87@gmail.com
Recibido el 29 de julio de 2015.
Aceptado para su publicación el 2 de diciembre de 2015.

