










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSanidad Militar
versión impresa ISSN 1887-8571
Sanid. Mil. vol.67 no.4 Madrid oct./dic. 2011
https://dx.doi.org/10.4321/S1887-85712011000500002
Gestión del riesgo en la transferencia de procesos productivos. Aplicación a la fabricación de comprimidos de oseltamivir en la pandemia de gripe A
Risk management in the transfer of manufacturing processes. Application to the manufacturing of oseltamivir tablets in the swine flu pandemic.
Juberías Sánchez A.1, Zamanillo Sainz A.2, Cabrera Merino JI.1, Verón Moros M.3, Urquía Grande MaL.4, Gonzalo Salado MaL.5
1Tcol. Farmacéutico. Centro Militar de Farmacia de la Defensa-Burgos. España.
2Tcol. Farmacéutico. Inspección General de Sanidad. Madrid. España.
3Lda. en Farmacia. Centro Militar de Farmacia de la Defensa. Madrid. España.
4Dra. en Farmacia. Centro Militar de Farmacia de la Defensa-Madrid. España.
5Cte. Farmacéutico. Centro Militar de Farmacia de la Defensa-Madrid. España.
Dirección para correspondencia
RESUMEN
Introducción: La gestión de riesgos aplicada a la industria farmacéutica, a través de su identificación, valoración y control, es una herramienta útil para garantizar la calidad del medicamento. La declaración por la Organización Mundial de la Salud en el año 2.009, de la pandemia provocada por el virus de la influenza H1N1, origina la necesidad de transformar en medicamento parte de las reservas estratégicas de fosfato de oseltamivir, pertenecientes al Ministerio de Sanidad y Política Social y a las Comunidades Autónomas. Debido a esta circunstancia, se dibuja, un nuevo escenario de actuación de los Servicios Farmacéuticos de la Defensa, bajo la tutela de la Agencia Española de Medicamentos y Productos Sanitarios.
Objetivo: La aplicación de los principios de gestión del riesgo, posibilitará una rápida adaptación de la tecnología necesaria para la fabricación de comprimidos de fosfato de oseltamivir. Se realiza la evaluación del riesgo asociado a las diversas fases del proceso y se establecen determinados parámetros de control sobre la calidad final del medicamento producido.
Material y Método: Se aplica el Análisis modal de fallos modales y efectos y su criticidad para establecer y evaluar los posibles riesgos del proceso productivo y se efectúan las correspondientes determinaciones analíticas referidas a la calidad del producto obtenido.
Resultados: Los riesgos asociados al proceso son establecidos y evaluados. La media del contenido de los comprimidos y del porcentaje de disolución de los mismos a los 20 minutos es de 101,9 % y 102,5% respectivamente, cumpliendo todos los lotes fabricados el ensayo de evaluación de la contaminación microbiológica. Los resultados obtenidos para los lotes en las variables objeto de validación del proceso (peso de los comprimidos y porcentajes de principio activo, de homogeneidad de la mezcla y de disolución) cumplen las especificaciones establecidas y no se aprecian diferencias significativas entre los diferentes lotes (p>0,05).
Conclusiones: Los resultados obtenidos corroboran el éxito de la transferencia del proceso productivo de comprimidos de fosfato de oseltamivir a las instalaciones y equipos disponibles, así como la consecución de un proceso robusto y repetitivo, que proporciona un medicamento ajustado a las especificaciones de calidad establecidas.
Palabras clave: Gestión del riesgo. Transferencia de procesos. Adaptación equipos. Antivirales. Fosfato de Oseltamivir.
SUMMARY
Introduction: Risks management applied to the pharmaceutical industry, through their identification, evaluation and control, is a useful tool to guarantee drug quality. The declaration of swine flu H1N1 pandemic in 2009 by the World Health Organization, resulted in the need to transform into tablets part of the strategic reserves of oseltamivir phosphate of the Ministry of Health and Social Policy and the Autonomous Communities. This situation led to a change in the activity of the Defense Pharmaceutical Service under the auspices of the Spanish Agency for Medicines and Health Products.
Objective: Applying the principles of risk management makes possible a fast adaptation of the technology to manufacture the oseltamivir phosphate tablets. We evaluated the risk associated to the different stages of the process and established particular control parameters of the final quality of the product.
Materials and Methods: We applied the modal analysis of failures and effects and their criticality to establish and evaluate possible risks of the manufacturing process and carried out the necessary lab tests to check the quality of the product. Results: The risks associated to the process are established and evaluated. The average tablet content and the dissolution percentage at 20 minutes is 101.9% and 102.5%, respectively. All batches passed the microbiological contamination control. The results obtained for all batches in the validating variables of the process (weight of the tablets, percentages of active principle, homogeneity of the mix and dissolution) fulfill the required specifications and there are not significant differences among the different batches (p>0,05).
Conclusions: The results confirm the successful transfer of the manufacturing process of the oseltamivir phosphate tablets to the facilities and equipment available, as well as the achievement of a robust and repetitive process that provides a drug fulfilling the required quality specifications.
Key words: Risk management, Process transfer, Equipment adaptation, Antivirals, Oseltamivir phosphate.
Introducción y antecedentes
Gestión de riesgos y producción farmacéutica
La gestión del riesgo se configura como una herramienta orientada a descubrir la probabilidad de aparición de un riesgo y a valorar la severidad de aquél; esto es, la capacidad que presenta el riesgo de producir un daño, mediante la aplicación de una determinada sistemática1.
La gestión de riesgos, aplicada a los Sistemas de Calidad en la industria farmacéutica, apunta a ser un valioso componente de soporte para valorar la eficacia de los mismos2,3. Su aplicación, es recogida en el ámbito de la Comisión Europea a través de la Guía de Normas de Correcta Fabricación de medicamentos y de medicamentos en investigación de la Unión Europea, en concreto, en su Anexo número 20, incorpora, a partir del 1 de marzo de 2008, la directriz ICH Q9 "Quality risk Management" dictada por la International Conference or Harmonisation of technicals requirements of pharmaceuticals of human use, de noviembre de 20054.
Este documento enfoca de modo sistemático la gestión de riesgos, consistente en su identificación, valoración y control, considerando su integración en el sistema de gestión de la calidad. Debido a ello, un enfoque adecuado de la gestión de riesgos garantizará una elevada calidad del medicamento destinado al paciente, proporcionando información eficaz para la toma de decisiones. Una gestión de riesgos soportada en fundamentos científicos, y orientada a la protección del paciente, se configura como fundamento básico del sistema5.
La necesidad de evaluar, de manera rápida y eficaz, la posibilidad de asignar una determinada producción farmacéutica a unos equipos e instalaciones preexistentes, con el fin de asegurar la mínima variabilidad del producto obtenido, inducen a utilizar la gestión de riesgos como herramienta útil para este fin, dada su implicación con la calidad del producto. De este modo se destacarán los puntos críticos de un proceso y los equipos e instalaciones asociados a los mismos.
Ante la alerta sanitaria originada por una posible pandemia de gripe, se dibuja un nuevo escenario de actuación de los Servicios Farmacéuticos de la Defensa, como el establecido por el contenido de los Protocolos de 7 de diciembre de 2005 y 5 de febrero de 2008, suscritos entre el Ministerio de Sanidad y Consumo (actualmente Sanidad y Política social) y el Ministerio de Defensa, de los que se derivan determinados cometidos relacionados con la producción y distribución farmacéutica, asignados al Ministerio de Defensa, previa autorización de la Agencia Española de Medicamentos y Productos Sanitarios.
Estos cometidos determinan los siguientes campos de trabajo y colaboración:
• Custodia de Oseltamivir fosfato, principio activo, y de los medicamentos Tamiflú® (Oseltamivir) y Relenza® (Zanamivir).
• Elaboración de solución y comprimidos de fosfato de Oseltamivir ante una posible pandemia provocada por virus de la influenza H5N1.
• Fabricación de medicamentos por causas excepcionales relacionadas con la salud pública o por demandas ocasionadas por conflictos y catástrofes.
• Fabricación de determinados medicamentos sin interés comercial.
• Fabricación de antídotos.
• Distribución de medicamentos en programas de cooperación y ayuda.
Tal y como podemos comprobar, los Servicios Farmacéuticos de la Defensa deberán orientar parte de su producción y distribución farmacéutica hacia nuevos medicamentos y sectores poblacionales. Para asumir correcta y rápidamente estos nuevos cometidos, se sugiere la necesidad de disponer de una herramienta eficaz que evalúe la capacidad de las instalaciones y equipos de producción farmacéutica.
Antecedentes regulatorios
La nueva actividad, relacionada con la salud pública, se sustenta en las siguientes normas y regulaciones:
La Ley 29/2006 de Garantías y Uso racional de los Medicamentos y Productos Sanitarios, permite a las Autoridades, ante necesidades excepcionales y urgentes de medicamentos; adoptar medidas especiales en relación con su fabricación, importación, distribución y dispensación (artículo 2.3); la concesión, por parte de la Agencia española de Medicamentos y Productos Sanitarios de autorizaciones especiales de comercialización de medicamentos ante circunstancias excepcionales (artículo 24); la constitución de un "Deposito estatal estratégico para emergencias y catástrofes o promover la fabricación y comercialización de medicamentos sin interés comercial" (Disposición adicional).
Por su parte, diversas disposiciones Comunitarias, promueven diferentes actuaciones, en determinadas circunstancias que afecten la salud pública, como la promoción de determinados medicamentos y tratamientos, la creación de reservas de antivirales6,7 que compensen el periodo de demora en la comercialización de una vacuna específica e incremente la sensación de seguridad en la población o el desarrollo de modelos público-privado de contramedidas para las que no existe un mercado natural8; ámbitos, todos ellos, en los cuales se encuadraría la producción de medicamentos por los Servicios Farmacéuticos de la Defensa.
Producción de antivirales
La declaración por la Organización Mundial de la Salud en el año 2009, de la pandemia provocada por el virus de la influenza H1N16, origina la necesidad de transformar en medicamento parte de las reservas estratégicas de fosfato de oseltamivir, materia prima, pertenecientes al Ministerio de Sanidad y Política Social y a las Comunidades Autónomas.
La preparación de una forma farmacéutica utilizable por toda la población, disponible en un periodo de tiempo aceptable y con una estabilidad suficiente, que permita su distribución y almacenamiento con las mayores garantías, orientan la fabricación hacia comprimidos ranurados de rápida disolución, con 30 mg de Oseltamivir base. Esta presentación permite abarcar toda la gama de tratamientos preventivos y curativos (dosis de 30, 45, 60 y 75 mg), de acuerdo a la edad del paciente. El proceso de producción de estos comprimidos fue desarrollado por la Pharmacie Centrale des Armées, laboratorio de producción farmacéutica perteneciente al Service de Santé des Armées de Francia.
Con el fin de posibilitar la transferencia tecnológica necesaria para la fabricación de este medicamento, por los Servicios Farmacéuticos de la Defensa, es necesaria la suscripción de sendos acuerdos entre los Ministerios de Defensa Español y Francés, así como entre el Ministerio de Sanidad y Política Social y la firma farmacéutica "Laboratorios Hoffman la Roche", para el procesado de la sustancia activa protegida por una patente.
Justificación del estudio
Proceso de producción
Tal y como se ha indicado, la presentación y composición del medicamento (comprimidos ranurados con 30 mg de Oseltamivir base), permite su administración a toda la población, sin necesidad de recurrir a "extensiones de línea", entendiendo como tales otras formas farmacéuticas, vías de administración y concentración de un medicamento ya autorizado, según define el artículo. 111.2 de la Ley 29/2006, de 26 de julio, de Garantías y uso racional de los medicamentos y productos sanitarios.
Dada la existencia de un compromiso de confidencialidad, referido al know-how del proceso de producción, incluido en los acuerdos establecidos entre los Ministerios de Defensa Francés y Español, se opta por proporcionar sólo la información necesaria, que justifique un conocimiento general del proceso, pero que permita considerar los elementos que determinen el éxito de una transferencia.
El diseño del medicamento a transferir permite la compresión directa de una mezcla pulverulenta con granulometría específica, que evita los inconvenientes de otros procesos industriales más complejos, como por ejemplo la granulación húmeda, esta particularidad le confiere las características de robustez y simplicidad9,10.
La composición relativa de la mezcla se muestra en la Tabla 111. Las operaciones realizadas (preparación previa, mezclado, tamizado y compresión) permiten la obtención de comprimidos cuyos parámetros determinantes de la calidad deberán mantener valores que permanezcan constantes y sometidos a especificación durante todo el proceso; estos valores, que se muestran en la Tabla 2, se recogen en CTD-Modulo 3 Oseltamivir PG 30 mg comprimido ranurado, depositado en la Agencia Francesa de medicamentos y productos sanitarios (AFSSAPS) y aceptado por Agencia Española de medicamentos y productos sanitarios (AEMPS).
Las operaciones de acondicionamiento primario se realizan en lámina blíster formada por Cloruro de polivinilo 250 micras-Cloruro de polivilideno 60 micras + Complejo de aluminio 0,25 micras, utilizando un formato con 10 comprimidos que permite obtener el máximo rendimiento de los equipos existentes.
Identificación del riesgo en procesos, instalaciones y equipos
Para cada una de las fases de la producción se procederá a identificar los riesgos asociados a las mismas. Estos riesgos tendrán su origen en las operaciones realizadas, en los equipos utilizados y en las instalaciones asociadas al proceso12.
El uso de equipos diferentes a los originarios, que realicen operaciones con incidencia sobre determinadas características físicas del producto, tales como los involucrados en procesos de tamizado, mezclado, compresión y termoformado, determinarán en gran medida, los riesgos relacionados con la transferencia del proceso.
Para definir los riesgos, y proceder a su valoración, debemos considerar las siguientes preguntas4:
• ¿Qué puede ir mal?
• ¿Qué probabilidad hay de que algo vaya mal?
• ¿Qué consecuencias tiene?
La identificación de riesgos, permitirá establecer aquellos elementos determinantes de la adaptación del proceso a las nuevas instalaciones y equipos, mediante el control de determinados parámetros, los cuales servirán, asimismo, para efectuar una evaluación de la calidad final del producto obtenido y proponer posibles acciones de mejora que redunden en un incremento de calidad y/o productividad12.
Una vez identificados los riesgos, deberán someterse a un análisis exhaustivo, determinando las posibles causas de su origen, asignándoles una valoración, lo que permitirá destacar aquellos riesgos más significativos, susceptibles de gestión, por presentar una mayor incidencia sobre la calidad, eficacia o seguridad del producto, configurando por tanto, aquellas etapas críticas, determinantes en la adaptación del proceso, así como los parámetros asociados a estas etapas que deberán ser controlados.
Objetivos
Consideramos que la aplicación de los principios de gestión del riesgo, posibilitará una adaptación más rápida de una determinada tecnología, para la fabricación de un medicamento, a unas instalaciones y equipos diferentes a los originales13. La obtención de un producto que satisfaga las especificaciones contenidas en el documento de referencia CTD parte 3, será indicativo del éxito de la transferencia realizada.
Con este fin, procederemos a la evaluación del riesgo asociado a los diversos elementos del proceso y su relación con los equipos e instalaciones que serán utilizados; se establecerán determinados parámetros de control, cuya medición permite conocer la adaptación del proceso a las nuevas instalaciones y proporciona información sobre la calidad del medicamento fabricado.
De manera previa, debemos demostrar la adaptación del proceso a sus nuevas condiciones de trabajo3, para ello se procede a una validación14,15 del mismo, consistente en una evaluación de la calidad del producto obtenido sobre cinco lotes inicialmente fabricados; consideramos que el proceso está validado si el producto obtenido en cada uno de los lotes se ajusta a los requerimientos establecidos (Tabla 2) y no existen diferencias significativas entre estos lotes objeto de la validación. El mantenimiento de las especificaciones iniciales a lo largo del tiempo, circunstancia recogida en los estudios de estabilidad, contribuirá a valorar de forma definitiva el resultado de la transferencia efectuada; pero, dadas las condiciones de emergencia en que se produjo la transferencia y comienzo de la producción, la Agencia Española de medicamentos y productos sanitarios, estableció la distribución del medicamento con indicación de la fecha de fabricación. Simultáneamente a la producción de los comprimidos antivirales, se iniciaron los estudios de estabilidad correspondientes; dichos estudios, en la fecha de redacción de este texto, permanecen abiertos con el fin de establecer el periodo de validez del medicamento.
Material y Métodos
Proceso de producción
El proceso de producción se compone de las etapas reflejadas en la Tabla 3, donde se también se recoge, los locales y equipos asignados a las mismas. Los equipos empleados se describen a continuación:
• Operaciones Previas. Equipo: Compactador/Tamizador/calibrador Hosokawa-Bepex empleado para proporcionar un determinado tamaño de partícula a la mezcla realizada.
• Mezclado.Equipo: Mezclador bicónico Glatt, utilizado para la obtención de una distribución homogénea de principio activo en el seno de la mezclas.
• Compresión. Equipo: Maquina de comprimir Kilian RTS 21 y 26 con herramienta punzón ranurado cóncavo circular 8 mm de diámetro.
• Acondicionado: Descripción: Termoformador Marchessini MB 421, estuchadora Marchessini MA 155-BA 100, control de peso dinámico continuo, encajadora automática Marchessini PS 510 y Prodec. El acondicionado primario, efectuado en blíster, debe proteger el comprimido contenido en su interior hasta su consumo. El acondicionado secundario proporciona prospecto y estuche con la información apropiada.
Análisis de riesgos
La Herramienta utilizada es el Análisis modal de fallos modales y efectos y su criticidad (AMFEC) propuesta por el anexo I.3 de la Directriz ICH Q9. Mediante esta herramienta se evalúan los fallos que pueden acontecer en el proceso y sus consecuencias o efectos sobre el producto. Estas consecuencias son valoradas en orden a su gravedad y probabilidad2.
Para la clasificación de riesgos de acuerdo a su gravedad se utiliza la categorización establecida por la Agencia Europea del Medicamento (EMEA) en su clasificación de retiradas16,17.
• Clase I Alta: Defectos de calidad que son potencialmente nocivos para la vida.
• Clase II Media: Defectos que pueden causar malestar o enfermedad sin ser clase I o afectan a la calidad del producto.
• Clase III Baja: Defectos que no significan peligro para la salud o afectan levemente a la calidad del producto.
Respecto a la clasificación de los riesgos según su probabilidad se emplea la siguiente, propuesta por la Asociación de Farmacéuticos de Industria (AEFI)17:
• Alta: Suceso prácticamente seguro.
• Media: Suceso esperable, ha sucedido con anterioridad y es de suponer que vuelva a producirse en el futuro si no se toman acciones adicionales para mitigar el riesgo.
• Baja: Suceso que no ha sucedido en el pasado y no se espera que ocurra en el futuro, pero teóricamente probable.
La significatividad o importancia del riesgo será superior cuanto más elevadas sean su gravedad y probabilidad.
Distribución granulométrica mezcla
Se utiliza el método descrito en la Real Farmacopea Española18. Se pesan 100 gramos de material pulverulento y situarlos en batería de tamices de luz de malla decreciente. Proporcionar 525 golpes (35 golpes/minuto, durante 15 minutos). Se determina el peso en tanto por ciento, de la sustancia retenida en cada tamiz.
Densidad no golpeada
Se utiliza el método descrito en la Real Farmacopea Española18. Se introduce en una probeta 50,0 gramos de material pulverulento. Se toma la medida del volumen aparente (sin sedimentar). El valor de la densidad vendrá determinado por el cociente entre 50 y el volumen medido.
Velocidad de disolución comprimidos
Se utiliza el método descrito en la Real Farmacopea Española19. Una muestra de seis comprimidos se somete a disolución en medio acuoso, introducida en una cubeta, mediante dispositivo de agitación. En periodos de tiempo determinados se efectúa una valoración del contenido de principio activo disuelto en el medio. Se utiliza equipo Vankel mod. 7000 con siete cubetas de 900 ml y agitación por paletas tipo 2 USP, junto a baño termostático. La velocidad de rotación de las paletas se fija a 50 rpm. La temperatura del baño se ajusta a 37oC. La determinación de principio activo disuelto se realiza mediante espectrofotómetro UV/vis Varian Cary con lectura a 240 nm y celda de cuarzo. Los periodos de medición son de 5, 10, 15, 20 y 45 minutos.
Uniformidad peso muestra comprimidos
Se utiliza el método descrito en la Real Farmacopea Española20. Se toman 20 comprimidos y se pesan uno a uno en balanza analítica (Metler Toledo modelo AT 100) Solamente dos comprimidos pueden desviarse más de un 7,5% del peso de referencia establecido y ninguno podrá desviarse más del 15 % de este peso.
Homogeneidad de la mezcla en sustancia activa
Se utiliza el método descrito en la Real Farmacopea Española21. Se toma una muestra en la superficie, interior y fondo de la masa pulverulenta a granel de la mezcla del lote fabricado. Se determina el contenido en sustancia activa en cada una de las muestras por cromatografía líquida de alta resolución (HPLC) que consiste en inyectar las muestras problemas frente a un estándar de farmacopea de concentración conocida y comparando los cromatogramas obtenidos determinamos la riqueza de las muestras en la sustancia activa. El cromatógrafo utilizado es un HP Agilent Mod. 1200 compuesto por bomba cuaternaria, inyector automático, detector de diodos y software Chem-Station con algoritmo de integración mejorado para tratamiento de datos. La columna es una C-8 desactivada para bases (Symmetry® C8 5 μm 4,6 x 250 mm WAT 054270) y la longitud de onda de 207 nm.
Por ser un método transferido por el laboratorio francés (Pharmacie Centrale des Armées), se realiza un desarrollo analítico y puesta a punto del mismo y se procede a su validación. Se optimizaron los parámetros cromatográficos, para escoger las mejores condiciones de fase móvil, flujo, volumen de inyección y parámetros de integración.
Ningún valor individual obtenido podrá separase más del 15% respecto al valor medio de las tres muestras obtenidas. En aquellas mezclas utilizadas para la validación del proceso se efectúa el muestreo en seis puntos.
Uniformidad contenido de la sustancia activa en el producto terminado
Se utiliza el método descrito en la Real Farmacopea Española21. Se pulverizan 20 comprimidos y de esta mezcla se toma una parte alícuota, cuya masa sea igual al peso de referencia del comprimido, efectuar la determinación del contenido de la sustancia activa por cromatografía líquida de alta resolución como se describe en el apartado referente a la evaluación de la "Homogeneidad de la mezcla en sustancia activa". Se determinan tres muestras de 20 comprimidos por lote. Ningún valor obtenido podrá desviarse más del 15% del valor medio de las muestras obtenidas.
Uniformidad de dosis
Se utiliza el método descrito en la Real Farmacopea Española21. Se analiza el contenido en sustancia activa de 10 comprimidos unitarios por lote, mediante la técnica analítica de cromatografía líquida de alta resolución como se describe en apartado referente a la evaluación de la "Homogeneidad de la mezcla en sustancia activa". Los valores individuales podrán desviarse un 15% del valor medio de las muestras obtenidas, y solamente dos de ellos podrán alcanzar un máximo del 25% de desviación.
Control microbiológico
Se utiliza el método descrito por la armonización de Farmacopeas (Europea, Americana y Japonesa)22,23: Recuento de microorganismos viables aerobios y la ausencia de microorganismos patógenos. Se añaden asépticamente 10 gramos de comprimidos pulverizados, a un frasco con 90 ml de solución de peptona tamponada pH=7. Se sigue el método de dilución en masa y recuento en placa. Se añaden a 1 ml de la muestra con el tampón 20 ml del medio adecuado para el cultivo en cada caso: TSA (Trypcase Soja Agar) para bacterias viables aerobias y Saboraud Dextrosa Agar para mohos y levaduras. Se incuban en estufa a 30-35oC el medio TSA y a 20-25oC el medio Saboraud (2 placas de cada). Se hace el recuento después de 5 dias. Para la detección de Escherichia coli se realiza una fase de enriquecimiento en TSB (Caldo de Caseina Soja) 24 h a 30-35oC y después en Caldo Mc Conkey 24h a 42-44oC, otra fase de aislamiento en Agar Mc Conkey 24h a 30-35oC con confirmación del microorganismo si hubiera alguna colonia sospechosa.
Los criterios de aceptación son los descritos en la farmacopea armonizada para las preparaciones de uso oral no acuosas25:
• <103 ufc/g de bacterias aerobias.
• <102 ufc/g de hongos.
• Ausencia de Escherichia coli en 1 g.
De manera previa al establecimiento de la metódica, se realiza una verificación ("suitability of the method")22,23. Para ello se inoculan un máximo de 100 ufc de cada cepa por separado (E. coli, S. aureus, P. aeruginosa y C. albicans) a los comprimidos de oseltamivir, comprobando su recuperación.
Ensayo estanqueidad blíster
Se determina mediante dos métodos diferentes que a continuación se describen, pertenecientes a la transferencia tecnológica realizada con el Ministerio de Defensa francés:
Determinación de la estanqueidad de envase primario blíster con solución de azul de metileno
Se introduce los blíster a ensayar en solución de azul de metileno al 1% en un recipiente estanco. Generar vacío de 750 mbar. Mantener este vacío durante 2 minutos. Restablecer la presión y mantener en reposo los blíster sumergidos en la solución de azul de metileno durante 2 minutos más. Observar que la solución de azul de metileno no se ha introducido en el interior del blíster manchando el comprimido.
Determinación de la estanqueidad con solución de Rodamina
Se introducen los blíster a ensayo en solución alcohólica de rodamina del 20%. Mantener sumergidos durante 30 minutos. La baja tensión superficial de la solución favorecerá su penetración por los poros o aberturas existentes. Observar que la solución de rodamina no se ha introducido en el interior del blíster manchando el comprimido.
Análisis estadístico
Se realiza un tratamiento estadístico mediante el programa informático SPSS versión 15 (Stadistical Package for the Social Science).Se calculan los datos descriptivos, diagramas de cajas y análisis de varianza con comparaciones múltiples de las variables utilizadas en la validación: Distribución peso, dosis por comprimido, homogeneidad mezclas y porcentaje de disolución del contenido en principio activo por comprimido en diferentes lotes.Para analizar la calidad global del proceso de producción, se comprueba la normalidad de la distribución de peso, densidad, contenido, porcentaje de disolución del contenido en principio activo por comprimido en los lotes producidos, realizándose un análisis de los datos descriptivos.
Resultados
El Análisis modal de fallos y efectos y su criticidad (AMFEC), aplicado al proceso, arroja los resultados reflejados en las tablas 4 y 5. En la Tabla 4 se relacionan las diferentes etapas del proceso, los equipos e instalaciones implicados y los riesgos asociados a las mismas, se resaltan en cursiva los riesgos directamente relacionados con la transferencia del proceso a equipos diferentes de los utilizados en su concepción y desarrollo inicial.
La Tabla 5 refleja los posibles efectos asociados a los riesgos, identificados previamente y una valoración de los mismos, basada en su gravedad (clase I, II, III) y peligrosidad (alta, media, baja) designadas en la tabla como (1, 2, 3) y (A, M, B) respectivamente. En esta misma tabla se proponen una serie de determinaciones, que permitan obtener información sobre el control de los riesgos establecidos. Se resaltan en cursiva los controles relacionados con los riesgos derivados de la transferencia del proceso, indicados en la Tabla 4.
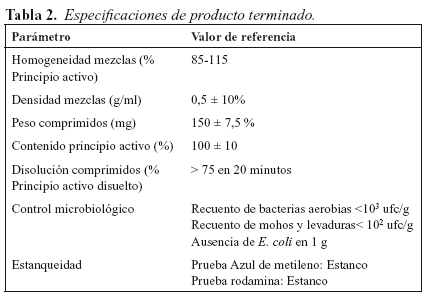
Los resultados correspondientes a los parámetros determinantes de la calidad del producto, en aquellos lotes sometidos a validación, se recogen en la Tabla 6 y Figura 1 (diagrama de cajas).
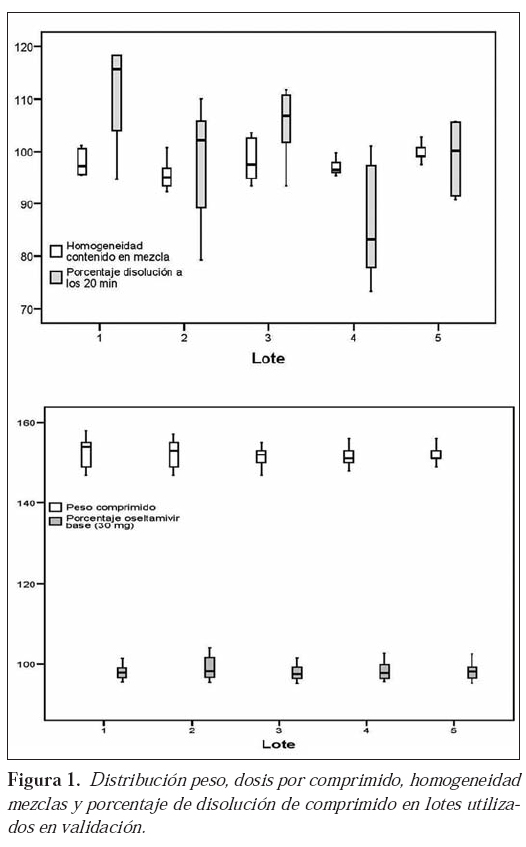
La distribución del tamaño de partícula de las mezclas, pertenecientes a los lotes objeto de validación, se recoge en la Figura 2, donde se pueden observar agrupamientos similares de partículas en los diferentes lotes. La calidad de toda la producción efectuada, mediante el estudio de los parámetros establecidos, se recoge en la Tabla 7.
Todos los lotes, presentaron ausencia de contaminación microbiológica (0 ufc), según los resultados obtenidos en el recuento de microorganismos aerobios, recuento de mohos-levaduras y ausencia de E. coli. El número de comprimidos procesados para esta determinación fueron 67 comprimidos por lote.
El envase primario (blíster) resultó estanco en la totalidad de las muestras evaluadas; se someten a este ensayo un total de 390 blíster.
Discusión
De acuerdo al contenido de la Tabla 4, se destacan los equipos y procesos clave asociados a los mismos, los cuales, determinan que la transferencia del proceso, a unos equipos e instalaciones diferentes de las utilizados en su concepción original, sea la adecuada y proporcione un producto de calidad contrastada, de acuerdo a los parámetros determinantes de la misma recogidos en este estudio.
Describimos a continuación, cada fase y equipo asociado considerados críticos a la hora de asumir la producción y establecemos una relación con los resultados obtenidos.
Fase de operaciones previas
Riesgo: La inadecuación del equipo/proceso dará lugar a mezclas con una distribución granulométrica no apta para la compresión. Esta distribución granulométrica se relaciona íntimamente con la densidad no golpeada de la mezcla, pues el tamaño de partícula condiciona el valor de este parámetro; así, partículas de reducido diámetro conducirán a valores de densidad elevados9. Valoración de resultados: Tal y como hemos indicado, en la figura 2 se establece una correlación entre la distribución granulométrica de las mezclas utilizadas en la validación del proceso; estas mezclas exhiben un comportamiento adecuado en la compresión; las cinco mezclas, objeto de validación, muestran proporciones similares para cada tamaño de partícula.
Fase de mezclado
Riesgo: Un equipo inadecuado o un uso incorrecto (insuficiente o elevado número de vueltas en la fase de mezclado), darán lugar a mezclas heterogéneas o comprimidos cuyo contenido en principio activo sea variable y se sitúe fuera de los márgenes de tolerancia9.
Valoración de resultados: Tanto las mezclas objeto de validación (Tabla 4), como los resultados correspondientes a la totalidad de la producción (Tabla 7) nos indican que las mezclas obtenidas fueron homogéneas y los comprimidos obtenidos también lo eran.
Fase de compresión
Riesgo: Un equipo inadecuado proporcionará comprimidos cuyo peso se sitúa fuera de los márgenes de tolerancia o cuya dureza influya en la consistencia del comprimido y/o en el perfil de disolución del principio activo25.
Valoración de resultados: Tanto la Tabla 4 como la Tabla 7, nos muestran que el proceso, tanto en su fase de validación, como de producción industrial dio lugar a comprimidos cuyas especificaciones se corresponden con las establecidas, en cuanto a homogeneidad del peso, dureza y velocidad de disolución.
Fase de acondicionamiento primario
Riesgo: El cierre de la lámina blíster debe ser estanco y no presentar fisuras ni alteraciones que expongan el medicamento contenido en su interior. La estanqueidad del envase será evaluada con el fin de valorar la adecuación del equipo a la operación26.
Valoración de resultados: Todos los controles de estanqueidad del blíster cumplen con los requerimientos especificados.
Control de la contaminación
Riesgo: Las diversas operaciones integrantes del proceso de producción ponen el producto en contacto con el ambiente y con los diversos equipos que son utilizados, una inadecuada higiene ambiental y de los materiales en contacto con el medicamento puede provocar el desarrollo y aparición de gérmenes en el producto terminado.
Valoración de resultados: La ausencia de este tipo de contaminación evidencia la existencia de unas adecuadas condiciones ambientales de trabajo así como unos protocolos de limpieza satisfactorios, aplicados a instalaciones y equipos.
Por lo tanto, y en base a los resultados obtenidos, serán parámetros determinantes de la calidad del producto obtenido y de la eficacia en la transferencia del proceso, los siguientes26:
• Densidad aparente de las mezclas.
• Peso homogéneo de los comprimidos.
• Uniformidad del contenido en principio activo de los comprimidos.
• Velocidad de disolución de comprimidos.
• Recuento de microorganismos.
• Estanqueidad de envase primario (blíster).
La adaptación del proceso a equipos e instalaciones se ve reflejada en los resultados obtenidos en los lotes utilizados en la validación. Los resultados reflejados en la tabla 4 muestran valores similares y acordes a las especificaciones establecidas para el producto, los diagramas de cajas de la distribución de las cuatro variables (homogeneidad de mezclas, uniformidad peso, contenido en principio activo y velocidad de disolución de comprimidos) en los lotes utilizados para la validación (Figura 1) muestran un análisis visual que sugiere que no hay modificaciones entre lotes en las cuatro variables mencionadas anteriormente. Esta suposición queda confirmada con el análisis de varianza, demostrándose la similitud entre los cinco lotes (p>0,05), no existiendo diferencias significativas en las comparaciones de las medias entre todos los lotes (p>0,05), realizado con la prueba de Bonferroni.
La homogeneidad de los lotes estudiados en la validación del proceso, así como la ausencia de diferencias significativas entre ellos, demuestra que el proceso se transfiere con éxito y que es robusto y repetitivo.
La calidad global del proceso, recogida en la Tabla 7, reafirma el éxito de la transferencia y adaptación del proceso a las instalaciones y equipos, al proporcionar resultados acordes con las especificaciones de producto para la totalidad de los lotes fabricados; además los resultados obtenidos muestran valores próximos entre sí, en los intervalos de la media, para un grado de confianza del 95 %, lo que manifiesta su fiabilidad27.
Conclusiones
El análisis de riesgos se configura como una herramienta adecuada para valorar la capacidad de adaptación de instalaciones y equipos a un determinado proceso; esta circunstancia es especialmente valorada en el ámbito de la Producción Farmacéutica Militar, si tomamos en consideración la creciente versatilidad exigida a la misma, tanto para atender las necesidades de las Fuerzas Armadas como en sus nuevos campos de actuación, tales como producción de antivirales, antídotos o medicamentos sin interés comercial, muchos de ellos destinados a la población civil.
En relación con la producción de comprimidos antivirales de fosfato de oseltamivir, la aplicación de esta metodología ha permitido establecer los diferentes riesgos relacionados con la transferencia del proceso, así como los controles necesarios para su adecuada gestión y evaluación de la calidad del producto obtenido, con la rapidez necesaria frente a una situación de emergencia, como la acontecida.
Los resultados obtenidos en el estudio de la calidad general de todos los lotes fabricados, corroboran el éxito en la transferencia del proceso productivo a las instalaciones y equipos disponibles, así como la consecución de un proceso robusto y repetitivo, que proporciona un medicamento ajustado a las especificaciones de calidad establecidas.
Después de la experiencia y resultados obtenidos en este nuevo campo de trabajo, destinado a satisfacer una demanda social en circunstancias muy especiales, se podría considerar a los Centros de Producción Farmacéutica del Ministerio de Defensa como una posible opción para la fabricación de ciertos medicamentos o antídotos demandados por la sociedad y que por restricciones de tiempo o costes, existencia de patentes o razones de oportunidad, precisaran la actuación de laboratorios estatales.
Bibliografía
1. Salazar Macian R. "Cualificación y validación: elementos básicos de la calidad y productividad". 2007:129. [ Links ]
2. Moheb M, Donghao R, Chi-wan Ch. "cGMP,s for the 21st Century and ICH Quality Initiatives". Pharmaceutical dosage forms: Tablets. Volume 3: Manufacture and process control. Third Edition. Informa Healthcare USA, Inc. 2008. Chap. 8 - 237:249. [ Links ]
3. García Bueno N, Giribets Parra E, González Gracia S, de Pablo Sedano M, Saumench Perramon L, Soler Ranzani L, Tébar Pérez A, Viola Demestre, C. "Transferencia tecnológica: presente y futuro. La nueva perspectiva de las ICH Q8/Q9/Q10". Industria Farmacéutica - Julio Agosto 2007- no 134:40-46. [ Links ]
4. EMEA (European Medicines Agency). International Conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use. "Quality Risk Management Q9". November 2005. [ Links ]
5. Guía de Normas de Correcta Fabricación de Medicamentos de Uso Humano y Veterinario. Anexo 20: Gestión de riesgos para la calidad. Adopción de la directriz ICH Q9. 1 marzo 2008:4. [ Links ]
6. Reddy D. "Responding to pandemic (H1N1) 2009 influenza: the role of oseltamivir", Journal Antimicrobial Chemotherapy 2010. Vol. 65 (Supplement 2):35-40. [ Links ]
7. Unión Europea. Comisión de las Comunidades Europeas. Documento COM (2004) 201. "sobre la planificación comunitaria de la preparación y la respuesta frente a pandemias de gripe". Bruselas, marzo 2004. [ Links ]
8. Unión Europea. Comisión de las Comunidades Europeas. Documento COM (2007) 0399. "Libro verde sobre la preparación frente a amenazas biológicas". Bruselas, julio 2007. [ Links ]
9. Vila Jato JL. "Tecnología farmacéutica. Volumen II: Formas Farmacéuticas". Editorial Síntesis. 1997:105. [ Links ]
10. Gohel MC, Pranav DJ. "A review of co-processed directly compressible excipients". J Pharm Pharmaceut Sci 2005 - 8(1):76-93. [ Links ]
11. Kibbe AH. "Handbook of Pharmaceutical Excipients". Third Edition. Pharmaceutical Press. London 2000. [ Links ]
12. Salazar Macian R. "Validación Industrial. Su aplicación a la industria farmacéutica y afines". 1999:28. [ Links ]
13. Canadell Heredia R, García Vidal E, Herrero Sas S, Llaja Villena J, Noguera Salvans L, Piñas Llagostera A, Puñal Peces D, Tardío Pérez E, Tebar Pérez A. "Gestión de desviaciones en un entorno ICH Q9/Q10". Industria Farmacéutica - Noviembre Diciembre 2007 - no 137:72-77. [ Links ]
14. Programa de Formación 2000 de AEFI (Asociación Española de Farmacéuticos de Industria): Validación de Equipos. Madrid 14-15 Diciembre 2000:6. [ Links ]
15. Ministerio de Sanidad y Consumo (Agencia española de medicamentos y productos sanitarios). Normas de correcta fabricación. Medicamentos de uso humano y veterinario. 3a Edición (2008). Anexo 15:275-281. [ Links ]
16. Salazar Macian R. "Cualificación y validación: elementos básicos de la calidad y productividad". 2007:63 y 127. [ Links ]
17. XXIV Symposium AEFI (Asociación Española de Farmacéuticos de Industria). Sección Centro. Grupo de trabajo de NCF. "Análisis de riesgos en acondicionamiento primario y secundario. Propuestas de controles en proceso". Córdoba 18 y 19 de noviembre de 2004. [ Links ]
18. Ministerio de Sanidad y Consumo. Agencia española de medicamentos y productos sanitarios. Real Farmacopea Española 3a Edición. Madrid 2005:266. [ Links ]
19. Ministerio de Sanidad y Consumo. Agencia española de medicamentos y productos sanitarios. Real Farmacopea Española 3a Edición. Madrid 2005:251-254. [ Links ]
20. Ministerio de Sanidad y Consumo. Agencia española de medicamentos y productos sanitarios. Real Farmacopea Española 3a Edición. Madrid 2005:257. [ Links ]
21. Ministerio de Sanidad y Consumo. Agencia española de medicamentos y productos sanitarios. Real Farmacopea Española 3a Edición. Madrid 2005:258. [ Links ]
22. EDQM (European Directorate for the Quality of Medicines & Healthcare). European Pharmacopoeia 6th Edition. June 2007. 2.6.12 "Microbiological examination of non-sterile products: Total viable aerobic counts" (01/2007:20612). [ Links ]
23. EDQM (European Directorate for the Quality of Medicines & Healthcare). European Pharmacopoeia 6th Edition. June 2007. 2.6.13 "Microbiological examination of non-sterile products: Test for specified micro-organism" (01/2007:20613). [ Links ]
24. EDQM (European Directorate for the Quality of Medicines & Healthcare). European Pharmacopoeia 6th Edition. June 2007. 5.1.4 "Microbiological quality of pharmaceutical preparations" (01/2007:50104). [ Links ]
25. Alderborn G, Frenning G. "Mechanical strength of tablets". Pharmaceutical dosage forms:Tablets. Volume 3: Manufacture and process control. Third Edition. Informa Healthcare USA, Inc. 2008. Chap. 7-207:236. [ Links ]
26. Gómez Alcaraz J, González Vázquez M, Tascón Herrero B, Alonso Navales A, Almazán Fernández T, Fernández Puerto J. "Validación de un proceso de fabricación de comprimidos por cambio de instalaciones". VII Congreso de Ciencias Farmacéuticas y XXVI Symposium de AEFI. Madrid 26 y 27 de octubre 2006. Libro de Ponencias: 480. [ Links ]
27. Pueyo Velasco J, Moliner Piqueras P, Sánchez Pallarés E, Sánchez Fraile E, Seguí Cosme JM, Rodríguez-Pina Burges R, Maganto Gómez J. "Validación del cambio de escala en la producción de comprimidos". VII Congreso de Ciencias Farmacéuticas y XXVI Symposium de AEFI. 26 y 27 de octubre 2006. Libro de Ponencias: 492. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
A. Juberías Sánchez.
Centro Militar de Farmacia de la Defensa.
Paseo de las Fuentecillas, s/n. 09001 Burgos.
ajubsan@oc.mde.es
Recibido: 28 de junio de 2010
Aceptado: 24 de marzo de 2011

