










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSanidad Militar
versión impresa ISSN 1887-8571
Sanid. Mil. vol.68 no.3 Madrid jul./sep. 2012
https://dx.doi.org/10.4321/S1887-85712012000300008
Absceso cerebral en paciente con Enfermedad de Rendu-Osler-Weber. Revisión del síndrome a partir de un caso ilustrativo
Brain abscess in patient with Rendu-Osler-Weber disease. Revision of the syndrome from an illustrative case
Arcos Sánchez C.1, Salinas Vela F.T.2, Echeandia Ajamil C.3, Olmedilla González M.N.4, Garrido Carrión A.5, Antón Barca R.6, Molina Foncea A.F.7
1 Cap. Médico. Servicio de Neurología.
2 Cap. Médico. Servicio de Radiodiagnóstico. Hospital General de la Defensa Orad y Gajías. Zaragoza. España.
3 Ldo. Medicina.
4 Cte. Médico.
5 Tcol. Médico.
6 Lda. Medicina. Servicio de Neurología.
7 Cte. Médico. Servicio de Neurocirugía.
Hospital Central de la Defensa Gómez Ulla.
Dirección para correspondencia
RESUMEN
Introducción: La Enfermedad de Rendu-Osler-Weber o telangiectasia hemorrágica hereditaria (THH) está considerada como una causa rara de ictus en paciente joven. Asimismo la presencia de malformaciones arteriovenosas pulmonares (MAVP) se ha relacionado con la aparición de abscesos cerebrales.
Método: Descripción del caso de una paciente de 52 años que, en el contexto de un ataque de migraña, presenta una pérdida de fuerza en hemicuerpo izquierdo acompañada de afectación de la sensibilidad tactoalgésica, progresivas. En la evolución observamos disminución del nivel de conciencia y signos de hipertensión intracraneal secundarios edema por la presencia de un absceso cerebral causado por un embolismo séptico paradójico debido a la existencia de MAVP.
Resultados: A través de la realización de una exhaustiva historia clínica se llega al diagnóstico de THH confirmado mediante los estudios genéticos, describiéndose una deleción completa, mediante técnica de MLPA (Multiplex Ligation-dependent Probe Amplification), de uno de los alelos del gen de la Endoglina (ENG).
Conclusión: La Enfermedad de Rendu-Osler-Weber, aunque infrecuente, debe considerarse en la etiología de los abscesos cerebrales en pacientes con historia de fistulas pulmonares. El seguimiento de los pacientes con THH es importante para minimizar el riesgo de aparición de procesos potencialmente graves. Aunque el diagnóstico sigue basándose en los criterios clínicos, los estudios moleculares pueden confirmar la presencia de mutaciones genéticas relacionadas con la enfermedad.
Palabras clave: Absceso cerebral. Ictus de causa rara. Malformaciones arteriovenosas. Síndrome de Rendu-Osler-Weber. Telangiectasia hemorrágica hereditaria.
SUMMARY
Introduction: Rendu-Osler-Weber syndrome also known as hereditary hemorrhagic telangiectasia (THH) is considered as a rare cause of stroke in young people. Also pulmonary arteriovenous malformations (MAVP) are related to brain abscess.
Method: We describe the case of a 52 years old female patient that, during a migraine attack, suffers a progressive onset of weakness in left body with numbness. In the evolution there appear signs of altered consciousness and intracranial hypertension, due to a cerebral abscess caused by a septic embolism as a manifestation of PAVM.
Results: Through the clinical history we reached the diagnosis of THH confirm by genetic studies, our patient shows, by means of MLPA (Multiplex Ligation-dependent Probe Amplification), a complete delection of one of the alelo of Endogline (ENG) gene.
Conclusions: Rendu-Osler-Weber disease, although its low prevalence, should be consider as a cause of brain abscesses in patients with history of pulmonary fistula. The follow of these patients with THH is very important to minimize the risk of fatal events. The diagnostic criteria are clinic but molecular studies can confirm the presence of genetic mutations related to this disease.
Key words: Brain abscess. Rare Stroke Syndromes. Arteriovenous malformations. Rendu-Osler-Weber syndrome. Hereditary Haemorrhagic Telangiectasia.
Introducción
El embolismo de origen no cardiaco, en este caso concreto de origen pulmonar secundario a Enfermedad de Rendu-Osler-Weber, se engloba dentro del heterogéneo conjunto de ictus de origen poco habitual, que representa aproximadamente un 7% de los ictus cerebrales isquémicos1, a pesar de lo cual es importante establecer el diagnóstico para poder hacer un tratamiento etiológico, pero sobre todo porque son la causa más frecuente de ictus en paciente joven. Se considera que este tipo de ictus tienen mejor pronóstico con una mortalidad inferior al 7% durante los primeros 30 días.
Método
Descripción del caso de una paciente de 52 años que acude a urgencias por presentar un cuadro de 72 horas de evolución consistente en cefalea pulsátil de localización bitemporal acompañada de nauseas y malestar epigástrico; déficit sensitivo en miembro superior izquierdo fluctuante, con extensión posterior a miembro inferior izquierdo; y pérdida de fuerza en brazo izquierdo. La paciente no presentaba patología intercurrente. Entre los antecedentes personales destaca que se trata de una paciente ex fumadora hace 17 años, con historia de migraña con aura con escasa respuesta al tratamiento, epistaxis de repetición y que había sido sometida a la embolización y reparación quirúrgica de fistulas arteriovenosas pulmonares hace 12 años, manteniendo desde entonces hipoxemia con saturación de oxígeno en torno al 90% y disnea de moderados esfuerzos. Tras reinterrogar a la paciente obtuvimos el importante antecedente familiar de padre diagnosticado de Enfermedad de Rendu-Osler-Weber.
En la exploración neurológica destaca déficit sensitivo y motor en extremidades izquierdas con hipoestesia tactoalgésica y fuerza 4/5, reflejos osteotendinosos presentes y simétricos y respuesta cutáneo plantar flexora bilateral, resto de exploración dentro de la normalidad. Ingresa en el Servicio de Neurología con el diagnóstico de Ictus isquémico de probable origen embólico. Se realiza un primer TC craneal en urgencias en el que no se evidencia lesión isquémica ni hemorrágica aguda.
En los 4 primeros días presenta empeoramiento de la focalidad motora con pérdida de fuerza progresiva hasta hemiplejia de extremidades izquierdas y aparecen crisis parciales simples en forma de clonias en miembro superior izquierdo, por lo que se inicia tratamiento con Levetiracetam endovenoso con buena respuesta al mismo.
Entre los estudios diagnósticos realizados destaca la realización de una angiografía pulmonar en la que se evidencian 3 fístulas arteriovenosas en lóbulo inferior derecho a nivel del segmento basal posterior que son embolizadas en el mismo procedimiento (Fig. 1).
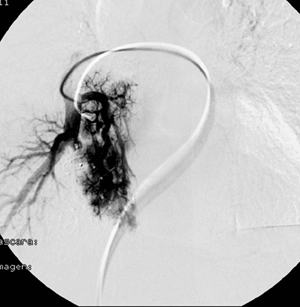
Figura 1. Angiografía pulmonar en la que se aprecia
malformación arteriovenosa pulmonar.
A los 6 días del ingreso presenta disminución brusca del nivel de conciencia con tendencia al sueño y con apertura ocular ante estímulos verbales, desorientación y localización ante estímulos dolorosos, se realiza nuevo TC craneal urgente en que se observa una imagen compatible con un absceso cerebral de localización frontoparietal izquierdo de 4.3cm de diámetro (Fig. 2). Por lo que, ante el empeoramiento clínico y, consultado con el Servicio de Neurocirugía, se decide realizar drenaje quirúrgico urgente (Fig. 3), que se consigue con éxito, y se inicia tratamiento antibiótico intravenoso. Tras la intervención quirúrgica se realizan controles sucesivos de neuroimagen a las 48 horas, a los 7 días, a los 14 días y los 21 días tras la intervención, observándose la disminución progresiva de la lesión descartándose la necesidad de reintervenir. La evolución clínica de la paciente es favorable presentando mejoría progresiva de la focalidad motora y sensitiva. La buena situación de la paciente hace posible el inicio de una rehabilitación precoz que hace que consiga la bipedestación y marcha independiente. Para completar el estudio, a nuestra paciente se le realizó una angioresonancia encefálica en la que no se evidenciaron malformaciones arteriovenosas cerebrales, así como una ecografía hepática en la que también se descartaron las lesiones a este nivel.
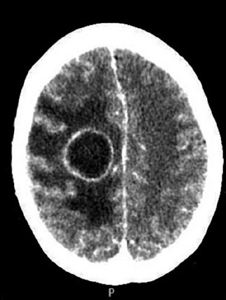
Figura 2. TC craneal, lesión hipodensa compatible
con absceso cerebral frontoparietal derecho de 4,3 cm de diámetro.
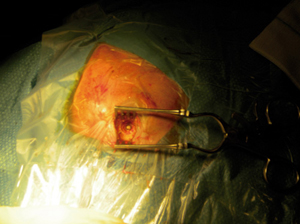
Figura 3. Craniectomía. Fotografía tomada durante la intervención
quirúrgica urgente de drenaje del absceso cerebral mediante trepano.
En cuanto al tratamiento elegido para nuestra paciente, se llevó a cabo mediante punción y drenaje ya que es menos agresivo, se puede obtener material para cultivo y confirmación diagnóstica asimismo se obtiene un alivio inmediato del aumento de la presión intracraneal, objetivo primordial en nuestro caso, y además puede realizarse en cualquier fase del absceso. En el estudio microbiológico del material obtenido con el drenaje del absceso se aisló Streptococcus Intermedius, una vez obtenido el resultado se suspendió antibioterapia empírica y se inició tratamiento antibiótico etiológico por vía parenteral durante 14 días con buena respuesta clínica y radiológica. En nuestra paciente pudimos observar clínicamente las cuatro etapas descritas en la formación de un absceso cerebral. Este calendario teórico lo pudimos ver in situ correlacionándose con las manifestaciones clínicas que presentó nuestra paciente a lo largo de la evolución del cuadro.
Las MAV tienden a crecer, esto es lo que ocurrió en nuestra paciente, hace 12 años ya se habían embolizado MAV de gran tamaño, las pequeñas, como se pudo observar en el nuevo ecocardiograma, habían crecido, y se hizo necesaria, una vez estabilizada la situación neurológica, una nueva embolización de las que cumplían criterios para este procedimiento intervencionista, llevado a cabo por el Servicio de Radiodiagnóstico de nuestro centro.
Discusión
El Síndrome de Rendu-Osler-Weber o telangiectasia hemorrágica hereditaria (THH) fue descrito por Sutton en 18642, aunque el primer informe de asociación entre telangiectasia y epistaxis hereditaria corresponde a Rendu en 18963, seguido por los de Osler y Weber4,5 en la última década del siglo XIX, en 1909 Hanes acuña el término THH para definir las características de la enfermedad6.
Se trata de un grupo de trastornos de herencia autosómica dominante que se estima que afectan, con una incidencia geográfica variable, de 1:2.000 a 1:10.000 individuos. Se han identificado dos genes en el cromosoma 9, que determinan mutaciones en el gen de la endoglina (ENG), y mutaciones en el gen ALK1, localizado en el cromosoma 12. Por lo que encontramos dos formas diferentes de la misma enfermedad, la variante THH1 se origina por mutaciones en el ENG, localizado en el brazo largo del cromosoma 9 (9q33-q34.1)7,8, y la forma THH2 está causada por mutaciones en el gen ALK1, localizado en el brazo largo del cromosoma 12 (12q11-q14)9. La endoglina es una glucoproteína de las membranas celulares de las células endoteliales, que se une al factor de crecimiento beta codificado por los genes ENG y ALK1, que modula la proliferación, migración, adhesión y organización de la matriz extracelular10,11. Ambos genes codifican una glicoproteína integral de membrana que se expresa en células del endotelio vascular y que actúa como receptor de superficie para el factor de crecimiento transformante β (TGF-β). Mutaciones en estos genes causan pues alteraciones en la angiogénesis que determinan las telangiectasias y las malformaciones arteriovenosas. La endoglina no se produce en cantidad suficiente o bien se trata de una proteína anómala que interfiere con el proceso de unión normal produciendo una displasia vascular en órganos específicos. Sin embargo, la existencia de grupos de familias con clínica compatible con la enfermedad pero sin mutaciones genéticas, sugiere que otro gen todavía no identificado podría ser la causa en estos casos de THH12. Además se ha descrito que las MAV pulmonares son más frecuentes en la THH113,14, y los desórdenes hepáticos son típicos del subtipo THH215-17. Aunque existen pacientes con THH2 y MAV pulmonares y pacientes con THH1 y manifestaciones hepáticas, por lo en ocasiones no se corresponde el diagnóstico genético con el comportamiento clínico16-18.
A pesar de estos avances el diagnóstico sigue siendo clínico, basado en la historia familiar y la presencia de malformaciones arteriovenosas, según los criterios diagnósticos propuestos por Plauchu et al.19 y Shovlin et al.20, estos son los criterios de Curaçao según los cuales el diagnóstico es definitivo si se cumplen tres de los criterios y posible en el caso de que solo se cumplan dos (Tabla 1).
Tabla 1. Criterios de Curaçao para el diagnóstico clínico. 
Para el diagnóstico molecular es necesario secuenciar las regiones codificantes completas de los genes ALK1 y ENG. El test genético no es positivo en el 100% de los pacientes con diagnóstico clínico de telangiectasia hemorrágica hereditaria.
Desde el punto de vista patológico se aprecia una alteración en el territorio capilar y la comunicación directa entre arteriolas y vénulas mediante fístulas, es pues una alteración vascular displásica multisitémica. En cuanto a las manifestaciones clínicas, la mayoría de los pacientes refieren epistaxis de repetición desde la infancia y asocian malformaciones arteriovenosas y/o múltiples telangiectasias cutáneas (fundamentalmente en cara y manos), tubo digestivo y cavidad oral, con tendencia al sangrado, con el consecuente déficit de hierro y anemia. Estas malformaciones, si no se tratan, son una causa infrecuente de ictus embólicos y abscesos cerebrales en familias con THH21. Las MAVP proporcionan una comunicación directa entre la circulación sistémica y el circuito pulmonar, lo que da lugar a un problema de oxigenación de la sangre arterial que conduce a la presencia de hipoxemia, además de la ausencia del filtrado capilar pulmonar, y la fragilidad capilar con el consiguiente riesgo de sangrado. La afectación neurológica es frecuente, estando presente hasta en un 30% de los casos, e incluye hemorragia intracraneal, cefalea, convulsiones, ictus isquémico y abscesos cerebrales, estos dos últimos cuadros siempre en relación con la presencia de MAVP, ya que el cortocircuito vascular permite la diseminación de trombos o émbolos sépticos hacia el cerebro. En cuanto a la cefalea, se ha descrito que hasta un 50% de los pacientes con THH presenta migraña, no estando clara la naturaleza de esta relación, pero es poco probable que se pueda atribuir a la presencia de malformaciones arteriovenosas cerebrales (MAVC), debido a su alta prevalencia. La presencia de MAVC se ha asociado a hemorragias subaracnoideas, parenquimatosas e intraventriculares y a hidrocefalia.
Según los estudios de Haitjema et al. las MAVP están presentes en el 33% de los pacientes con THH y se han podido observar eventos neurológicos graves hasta en un 40% de los pacientes con MAVP y THH22. Si se estudia a los pacientes con MAVP un 60% de estos presentan THH21. También se ha descrito que estas MAV tienden a aumentar su tamaño con el tiempo23. Las MAVP mayores de 3mm de diámetro precisan embolización para evitar las complicaciones descritas, así como el potencial riesgo de hemorragia pulmonar24. Las que son mayores de 1cm se pueden tratar mediante embolización, cirugía y/o radiocirugía25.
Para Lee et al. la ecocardiografía transtorácica es un método muy sensible para el screening de MAVP, detectando incluso las de menos de 3mm de diámetro26, por lo que sería la técnica de elección para completar el diagnóstico, que como ya se ha mencionado, sigue basándose en los criterios clínicos. El Doppler neurovascular también es útil para la detección de fístulas pulmonares mediante el test de infusión de burbujas27. La evaluación sistémica incluiría tomografía computerizada, resonancia magnética cerebral y ecografía hepática. Por último la profilaxis antibiótica está indicada en estos pacientes en todos los procedimientos dentales, endoscópicos y quirúrgicos para reducir el riesgo de abscesos cerebrales secundarios a embolismos paradójicos.
En cuanto a la etiopatogenia de los abscesos cerebrales, hasta 1980 el origen de un absceso cerebral era la propagación por contigüidad desde infecciones de senos paranasales o del oído, pero ha mejorado el tratamiento de estos cuadros por lo que, en la actualidad, la diseminación hemática es la causa más frecuente28. Los abscesos cerebrales ocurren en 1 de cada 100.000 personas aproximadamente, es una patología que se presenta fundamentalmente entre la segunda y la cuarta década de la vida y que es más frecuente en el sexo masculino. Se producen cuando los gérmenes alcanzan el parénquima cerebral fundamentalmente por tres vías: por inoculación directa o fístulas, en el caso de traumatismos o cirugía; por una infección en contigüidad (por invasión directa o por tromboflebitis de las venas emisarias); o por embolismos sépticos procedentes de infecciones a distancia. Como ya hemos mencionado la causa más frecuente es por propagación hemática. Los abscesos cerebrales en estos casos pueden ser múltiples hasta en un 10-15% de las ocasiones. El tórax suele es el punto de origen bien por abscesos pulmonares, bronquiectasias o empiema, en niños por cardiopatías cianóticas congénitas, sobre todo la tetralogía de Fallot (ya que los pacientes con comunicación izquierda-derecha pierden el efecto filtrante de los pulmones), o bien por MAVP, en este último caso aproximadamente el 50% de los pacientes presentan Síndrome de Rendu-Osler-Weber y, de estos, hasta un 5% presentarán abscesos cerebrales. Los abscesos cerebrales asociados a MAVP en pacientes con THH, revisten particular interés debido a la prevalencia de la enfermedad, por lo que debe sospecharse en todo paciente con absceso cerebral cuando los focos sépticos habituales se han descartado y en cualquier caso de MAVP con focalidad neurológica acompañante29. El diagnóstico fundamental se basa en las pruebas de neuroimagen que determinan el tamaño, la localización, la fase evolutiva, permiten planificar las opciones terapéuticas así como el seguimiento posterior de la lesión (se recomienda repetir el estudio de neuroimagen cada 1 ó 2 semanas aunque no exista deterioro neurológico). El aspecto típico es el de una lesión hipodensa rodeada de un anillo que realza tras la administración de contraste, se evalúa también el edema, la presencia de hidrocefalia o el desplazamiento de la línea media. Se describen 4 fases evolutivas, en la primera, de 2 a 3 días de duración, se produce una zona isquémica en el lugar de entrada del germen, con inflamación y desarrollo de un infiltrado leucocitario de células polimorfonucleares rodeado de edema vasogénico, es lo que vemos en los primeros estudios de neuroimagen realizados en las fases más precoces. En la segunda fase, de 4 a 9 días de duración, se produce un aumento de la zona necrótica y pus que rodea el infiltrado. En la tercera fase, entre el décimo y el decimotercer día, se delimita la zona purulenta y aparece una neovascularización rodeando el proceso. Y en la cuarta y última, a partir del día 14, el absceso se rodea de una capsula gliótica. La mortalidad de los abscesos cerebrales en pacientes con THH se sitúa alrededor del 10%, estas cifras de mortalidad han descendido debido al diagnóstico precoz de la enfermedad, el manejo adecuado de las MAVP y la profilaxis antibiótica llevada a cabo en estos pacientes30.
Las crisis epilépticas descritas, se engloban dentro del grupo de epilepsia aguda sintomática, tambien denominadas crisis reactivas, secundarias a una determinada situación. Se ha descrito que entre el 5 y el 10% de los pacientes con ictus presentan crisis precoces, estas suelen presentarse en la primera semana tras el evento vascular y generalmente se caracterizan por ser aisladas, breves y autolimitadas, suelen relacionarse, como en nuestro caso, con la topografía de la lesión siendo más frecuente en las de localización cortical parietotemporal31.
Resultados
La paciente cumplía tres criterios clínicos para el diagnóstico de THH según los criterios de Curaçao, la presencia de epistaxis, las MAVP y el antecedente familiar de padre diagnosticado de THH, con lo cual el diagnóstico era definitivo.
A pesar de esto, para completar el estudio, se realizó el diagnóstico molecular, con la colaboración del Servicio de Genética, mediante la secuenciación de las regiones codificantes completas de los genes ALK1 y ENG, que, como ya se ha mencionado no es positivo en el 100% de los casos de los pacientes con diagnóstico clínico de THH.
La muestra fue enviada al Centro de Investigaciones Biológicas del Consejo Superior de Investigaciones Científicas donde se realizó el estudio evidenciándose una deleción completa, mediante técnica de amplificación dependiente de ligasa de múltiples sondas, en inglés Multiplex Ligation-dependent Probe Amplification, de uno de los alelos del gen ENG, lo que confirma el diagnóstico molecular.
Conclusiones
La enfermedad de Rendu-Olser-Weber o THH es una enfermedad sistémica poco frecuente, con un amplio espectro clínico y con un riesgo importante de desarrollar complicaciones neurológicas; que la presencia de MAVP en los pacientes con THH puede hacer que émbolos, tanto trombóticos como sépticos, sobrepasen la barrera pulmonar dando lugar a fenómenos neurológicos de carácter embólico como abscesos cerebrales, ictus embólicos o ataques isquémicos transitorios; y que para reducir el riesgo de fenómenos embólicos, como el descrito, todas las MAV pulmonares mayores de 3 mm deben ser embolizadas, siendo preciso realizar seguimiento mediante ecocardiogramas transtorácicos periódicos ya que las MAVP tienden a crecer con el tiempo, necesitando el paciente nuevas embolizaciones.
Bibliografía
1. Benavente O. Ictus de origen poco habitual. Rev Neurol 2003; 36: 64-7. [ Links ]
2. Sutton HG. Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system. Medical Mirror 1864; 1: 679-81. [ Links ]
3. Rendu H. Epistaxis repetees chez un sujet porteur de petits angiomes cutanes et muquez. Gazette des Hopitaux Civils et Militaires (Paris) 1896; 135: 1322-3. [ Links ]
4. Osler W. On a family form of recurring epistaxis, associated with multiple telangiectases of the skin and mucous membranes. Bulletin of the Jonh Hopkins Hospital 1901; 12: 333-7. [ Links ]
5. Weber F. Multiple hereditary developmental angiomata (telangiectases) of the skin and mocous membranes associated with recurring haemorrhages. Lancet 1907; ii: 160-2. [ Links ]
6. Hanes FM. Multiple hereditary telangiectases causing hemorrhage (hereditary hemorrhagic telangiectasias). Bulletin of the John Hopkins Hospital 1909; 20(216): 63-73. [ Links ]
7. McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA, et al. A disease locus for hereditary haemorrhagic telangiectases maps to chromosome 9q33-34. Nat Genet 1994; 6: 197-204. [ Links ]
8. Shovlin CL, Hughes JM, Tuddenham EG, Temperley I, Perembelon YF, Scott J, et al. A gene for hereditary haemorrhagic telangiectasias maps to chromosome 9q3. Nat Genet 1994; 6: 205-9. [ Links ]
9. Johnson DW, Berg JN, Gallione CJ, McAllister KA, Warner JP, Hembold EA, et al. A second locus for hereditary haemorrhagic telangiectasias maps to chromosome 12. Genome Res 1995; 5: 21-8. [ Links ]
10. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-b type 1 receptors. EMBO J 2020; 21: 1743-53. [ Links ]
11. Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al. Activin receptor-like kinase (ALK) 1 as an antagonistic mediator of lateral TGF-b/ ALK5 signaling. Mol Cell 2003; 12: 817-28. [ Links ]
12. Wallace G, Shovlin C. A hereditary haemorrhagic telangiectasias family with pulmonary involment is unliked to the known HHT genes, endoglin and ALK-1. Thorax 2000; 55: 685-90. [ Links ]
13. Berg J, Guttmacher A, Marchuk D, Porteous M. Clinical heterogeneity in hereditary haemorrhagic telangiectasia: are pulmonary arteriovenous malformation more common in families linked to endoglin? J Med Genet 1996; 33: 256-7. [ Links ]
14. Heutink P, Haitjema T, Breedveld G, Janssen B, Sandkuijl L, Bontekoe C, et al. Linkage of hereditary haemorrhagic telangiectasia to chromosome 9q34 and evidence for locus heterogeneity. J Med Genet 1994; 31: 933-6. [ Links ]
15. Buscarini E, Buscarini L, Danesino C, Pinatanida M, Civardi G, Quaretti P, et al. Hepatic vascular malformations in hereditary haemorrhagic telangiectasia: Doppler sonographic screening in a large family. J Hepato 1997; 26: 111-8. [ Links ]
16. McDonals J, Miller F, Hallam S, Nelson L, Marchuk D, Ward K. Clinical manifestations in a large hereditary haemorrhagic telangiectasia type 2 kindred. Am J Med Genet 2000; 93: 320-7. [ Links ]
17. Olivieri C, Mira E, Delu G, Pagella F, Zambelli A, Malvezzi L, et al. Identification of 13 new mutations in the ACVRL1 gene in a group of 52 unselected Italian patients affected by hereditary haemorrhagic telangiectasia. J Med Genet 2002; 39: E39. [ Links ]
18. Abdalla S, Geisthoff U, Bonneau D, Plauchu H, McDonald J, Kennedy S, et al. Visceral manifestations in hereditary haemorrhagic telangiectasia type 2. J Me Genet 2003; 40: 494-502. [ Links ]
19. Plauchu H, de Chadaverian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32: 291-7. [ Links ]
20. Shovlin CL, Guttmacher AE, Buscarani E, Fuaghnan ME, Hyland RH, Westermann CJ et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Genet 2000; 91: 66-7. [ Links ]
21. Yoong JKC, Htoo MM, Jeyaseelan V, Ng DCC. Hereditary haemorrhagic telangiectasia with pulmonary arteriovenous malformations: a treatable cause of thromboembolic cerebral events. Singapore Med J 2004; 45(7): 334-6. [ Links ]
22. Haitjema T, Westermann CJ, Overtoom TT, Timmer R, Disch F, Mauser H, et al. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease): new insights in pathogenesis, complications and treatment. Arch Intern Med 1996; 156: 714-9. [ Links ]
23. White RI Jr. Pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia: embolotherapy using balloons and coils. Arch Intern Med 1996; 156: 2627-8. [ Links ]
24. Moussouttas M, Fayad P, Rosenblatt M, Hashimoto M, Pollak J, Henderson K, et al. Pulmonary arteriovenous malformations: cerebral ischemia and neurologic manifestations. Neurology 2000; 55: 959-64. [ Links ]
25. Fulbright RK, Chaloupka JC, Putman CM, Sze GK, Merrian MM, Lee GK, et al . MR of hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. Am J Neuroradiol 1994; 19: 477-84. [ Links ]
26. Lee WL, Graham AF, Pugash RA, Hutchison SJ, Grande P, Hyland RH, et al. Contrast echocardiography remains positive after treatment of pulmonary arteriovenous malformations. Chest 2003; 123: 351-8. [ Links ]
27. Más G, Sola D, García MA, Riba I, Hurtado R, Lopez N. Enfermedad de Rendu-Osler-Weber como causa infrecuente de ictus. Rev Neurol 2008; 46(5): 280-1. [ Links ]
28. Tonon E, Scotton P, et al. Brain abscess: clinical aspects of 100 patients. Int J Infect Dis 2006; 10(2): 103-9. [ Links ]
29. Shimokawa S, Koga H, Nakashima S, Anegawa S, Hayashi T. Multiple brain abscesses associated with Rendu-Osler-Weber disease: report of two cases. No To Shinkei 2001; 53(8): 775-9. [ Links ]
30. Rivero-Garvía M, Boto GR, Pérz-Zamarrón A, Alonso-Lera P, Zimman H, Saldaña CJ. Absceso cerebral asociado a la enfermedad de Rendu-Osler-Weber. Rev Neurol 2006; 43: 311-2. [ Links ]
31. Mauri-Llerda JA, Tejero-Juste C, Escalza-Cortina I, Jericó I, MoralesAsín F. Epilepsias agudas sintomáticas. Rev Neurol 2000; 31(8): 770-774. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Carolina Arcos Sánchez.
Servicio de Neurología,
Hospital General de la Defensa Orad y Gajías.
Vía Ibérica, 1,
50009, Zaragoza. España.
E-mail: carcsan@fn.mde.es
Recibido: 9 de abril de 2012
Aceptado: 11 de julio de 2012

