










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
versão On-line ISSN 2173-2345versão impressa ISSN 1889-836X
Rev Osteoporos Metab Miner vol.7 no.2 Madrid Abr./Jun. 2015
https://dx.doi.org/10.4321/S1889-836X2015000200004
Gitelman syndrome and chondrocalcinosis. A clinical case review
Síndrome de Gitelman y condrocalcinosis. Revisión de un caso clínico
Rosselló Aubach Ll.1, Vélez Cedeño V.K.2, Montalà Palou N.1, Conde Seijas M.1 and Palliso Folch F.1
1 Dirección Clínica del Aparato Locomotor - Hospital de Santa María de Lleida
2 CAP Les Borges Blanques - Lleida
SUMMARY
Gitelman syndrome is a tubulopathy of autosomal recessive inheritance which presents with, among other manifestations, hypomagnesemia and hypocalciuria. We present the case of a woman of 68 years of age who came for a consultation due to arthritis in the large joints, in the absence of other symptomology. The X-ray study showed deposits of calcium pyrophosphate in the knees, pubic symphysis and other joints. Blood tests revealed hypomagnesemia and hypocalciuria compatible with Gitelman syndrome, which was confirmed following a genetic study.
Key words: Gitelman syndrome, chondrocalcinosis, hypomagnesemia.
RESUMEN
El síndrome de Gitelman es una tubulopatía de herencia autosómica recesiva que cursa, entre otras manifestaciones, con hipomagnesemia e hipocalciuria. Se presenta el caso de una paciente mujer de 68 años que acudió a consulta por artritis de grandes articulaciones en ausencia de otra sintomatología. En el estudio radiológico se observaron depósitos de pirofosfato cálcico en rodillas, sínfisis púbica y otras articulaciones. En la analítica destacaba hipomagnesemia e hipocalciuria compatibles con síndrome de Gitelman que se confirmó tras estudio genético.
Palabras clave: síndrome de Gitelman, condrocalcinosis, hipomagnesemia.
Introduction
Gitelman syndrome is a disease transmitted by recessive autosomal inheritance, and is caused by mutations in the gene SLC12A3, located in the 16q13 chromosome, which codes for the synthesis of the Na+-Cl- cotransporter of the distal convoluted tubule [1], which produces a defect in the reabsorption of sodium. This increase in the loss of salt, in turn causes a moderate volume depletion which activates the renin-angiotensin-aldosterone system [2]. It is a tubulopathy characterised by hypomagnesemia, hypopotassemia with metabolic alkalosis and hypocalciuria. In most cases it manifests itself in adolescence or in adulthood and follows a more benign course than what is known as Bartter syndrome [3]. Most patients have low or normal arterial tension and may present with signs of volume depletion [4]. Their levels of urinary prostaglandin E2 are normal. It is important to emphasise that the severity of the symptoms is not related to the genotype pattern, nor is there a correlation with the laboratory test results in these patients. The differential diagnosis should be carried out with diuretic or laxative abuse and with patients with chronic emetic syndrome [5-6]. In spite of the fact that the association between Gitelman syndrome and chondrocalcinosis has already been known for some years, only in rare cases are chondrocalcinosis and hypomagnesemia presented together, such as occurred in our patient, due to the accumulation of calcium pyrophosphate crystals in the joints stimulated by the hypomagnesemia.
Clinical case
A female patient, 68 years of age, with no pathological history of interest, who came for a consultation due to repeated episodes of pain and inflammation in both knees, attributed until then to a degenerative process, and which improved with non-steroidal anti-inflammatories. During the last two years she also had pain in both wrists and cervical spine of a mechanical nature. She said she had not suffered episodes of diarrhoea or vomiting, did not consume diuretics or any other type of pharmaceutical drugs.
The examination showed a patient in a generally good state of health, normohydrated, with blood pressure of 120/80 mmHg. The rest of the examination showed pain and flexion/extension limitation in the right knee with positive meniscal manoeuvres, without signs of leaking joints. The hands showed degenerative signs in the distal interphalangeal joints suggestive of Heberden's nodes.
In the analyses, the haemogram and formula were normal. The biochemical analysis showed the following results: urea, 37 mg/dl; creatinine, 0.71 mg/dl; glomerular filtrate, >60 mL/min/1.73m; total calcium, 9.45 mg/dl; inorganic phosphate, 3.51 mg/dl; alkaline phosphatase, 56 U/L; sodium(Na), 140 mEq/l; potassium (K), 3.4 mEq/l; TSH, 3.45 mUL; blood PTH, 2.9 pmol/L (1.6-6.9); 25-hydroxicolecalciferol, 30.9 ng/ml (30-100); bone alkaline phosphatase, 9.7 ug/L; magnesium (Mg), 0,54 mmol/L (0,66-0,99). In urine at 24 hours: negative proteinuria; calciuria, 69.56 mg (100-250); phosphaturia, 588.30 mg; Mg, 1.31 mg/dL (1.7-5.7); phosphate in the first urine of the day, 15.9 mg/dL (40-136). The acute phase reactants, rheumatoid factor, anti-citrullinated antibody and antinuclear antibodies (ANA, anti-ENA) were normal or negative.
The X-ray study showed calcification in the menisci of both knees with additional degenerative signs (Figure 1), of the pubic symphysis, of both carpi, in the hyaline coxofemoral cartilage, as well as in the metatarsophalangeal joint in the big toe of both feet (Figure 2).
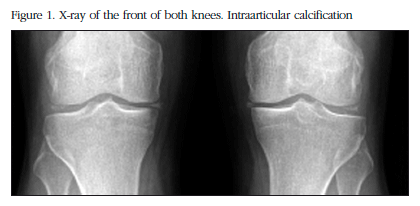
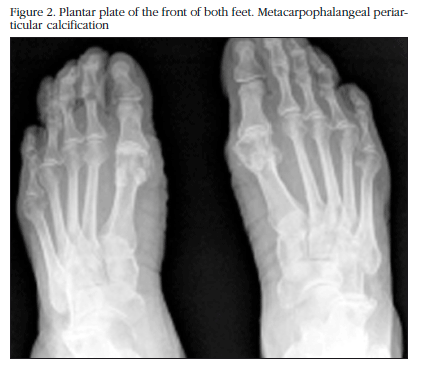
The nuclear magnetic resonance (NMR) of the right knee showed severe degenerative signs of patellofemoral, and internal and external tibiofemoral osteoarthritis with degenerative rupture of both menisci.
A molecular genetic study was requested using PCR amplification and sequencing of the SCL112A3 gene, detecting homozygosis of the c2576T>C(p.L859P) mutation in the exon of this gene and which confirmed the diagnosis of Gitelman syndrome. The treatment consisted of oral supplements of magnesium at variable doses depending on the results of the monitoring analysis, and 0.5 mg colchicine a day to avoid episodes of pseudogout which the patent was suffering.
Discussion
Gitelman syndrome was described by this author in 1966. It is an autosomal recessive hereditary disease resulting from the mutation in the long arm of chromosome 16 in which the SLC12A3 gene which codes for the thiazide-sensitive Na-Cl cotransporter in the distal tubule is affected. Its incidence is one case in every 40,000 people [7].
In most cases the symptoms do not appear before the age of seven, and the disease is generally diagnosed during adolescence or adulthood with very light symptoms,in some cases even being asymptomatic, and whose definitive diagnosis has to be made through a genetic study, as with our patient [8].
The physiopathology of Gitelman syndrome is the disturbance of the function of the thiazide-sensitive ClNa cotransporter (TSC) which results in the tubular reabsorption of chloride and sodium in the distal nephron, causing a loss of salt and water, with the consequent hypovolemia. The reduction in vascular volume activates the renin-angiotensin system, promoting an increase in the concentrations of renin and aldosterone. This, in turn, facilitates in the cortical collector duct an increase in the reabsorption of sodium in the apical membrane and an activation of the Na+-K+-ATPase in the basolateral membrane. The increase in the concentration of aldosterone stimulates the H+-ATPase in the cortical and medullar collector ducts, causing an increase in the secretion of H+ in the apical membrane. At the same time, the urinary secretion of potassium is increased due to the increase in the activity in in the basolateral membrane of the Na+-K+-ATPase. All this fosters the appearance of hypopotassemia alkalosis. The low intracellular content of sodium raises the tubular reabsorption of calcium through the activation of the Na+/Ca+ basolateral exchange, resulting in hypocalciuria. The magnesuria is increased by activating the Mg2+/Na+ exchange, given the existence of a negative transepithelial potential, which leads to the appearance of hypomagnesemia [9].
With regard to the renal function, a reduction in the renal tubular threshold for the reabsorption of magnesium without affectation of TmMg2 is confirmed. These data are compatible with the fact that most of the filtered magnesium is reabsorbed in the thick ascending limb of the loop of Henle, and that the distal tubule only reabsorbs around 5% of the filtered magnesium.The mechanisms for concentration and acidification are intact. In the hydrosaline overload the distal absorption of chloride and sodium are reduced [10].
The molecular mechanisms which link the hypomagnesemia to the chondrocalcinosis are not fully understood. While it is known that magnesium is a cofactor for many pyrophosphatases, such as alkaline phosphatase, which allows, in turn, the conversion of inorganic pyrophosphate into orthophosphate. The magnesium also increases the solubility of the crystals of calcium pyrophosphate. In states of hypomagnesemia this solubility of the calcium pyrophosphatase is changed, and the precipitation takes place of the crystals in the joints producing a crisis of pseudogout and also reduces the natural dissolution of these pyrophosphate crystals [11,12].
In Gitelman syndrome the patients are frequently asymptomatic, except for the appearance of recurrent episodes of muscular weakness and tetany, which may be accompanied by abdominal pain, vomiting and fever. The intervals of apparent health may be very prolonged and the diagnosis is not usually established until adulthood. However, almost half of the patients present lesser symptoms such as an appetite for salt, fatigue, muscle weakness, general aching, dizziness, nocturia and polydipsia [6,10].
Our patient had presented different episodes of pain and inflammation of the joints, especially in both knees, although due to the fact that secondary evolved osteoarthritis was probably added to the chondrocalcinosis and degenerative rupture of both menisci in the right knee, its association with Gitelman syndrome was not suspected until low levels of magnesium in the blood and of calcium in 24 hour urine were observed, which was subsequently confirmed by the genetic study.
In the treatment of Gitelman syndrome, the efficacy of the administration of salts of Mg exclusively (preferably MgCl, which compensates for the loss of both Mg and Cl in the urine) has been demonstrated, with normalisation of the biochemical parameters and clinical remission. A correction of the hypopotassemia is performed occasionally with the administration of potassium salts.
Indomethacin or the potassium-sparing diuretics (spironolactone or amiloride) are reserved for the most refractory cases.
With the presentation of this case we consider the determination of levels of magnesium ions in the blood, as well as calcium and magnesium in 24 hour urine, to be important in all those patients with chondrocalcinosis and in those in whom hypomagnesemia is suspected, in order to exclude Gitelman syndrome.
![]() Correspondence:
Correspondence:
Lluís Rosselló Aubach
Hospital de Santa María de Lleida
Dirección Clínica del Aparato Locomotor
Avda. Rovira Roure, 44
25198 Lleida (Spain)
e-mail: lrosello@gss.scs.es
Date of receipt: 06/05/2015
Date of acceptance: 19/06/2015
Bibliography
1. Konrad M, Weber S. Recent advances in molecular genetics of hereditary magnesium-losing disorders. J Am Soc Nephrol 2003;14:249-60. [ Links ]
2. De Jong JC, Van Der Vliet WA, Van Den Heuvel L, Willems PH, Knoers NV, Bindels RJ. Functional expression of mutations in the human Na-Cl cotransporter: Evidence for impaired routing mechanism in Gitelman's syndrome. J Am Soc Nephrol 2002;13:1442-8. [ Links ]
3. Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, et al. Gitelman's variant of Bartter's syndrome, in hereditary hypokalemic alkalosis, is caused by mutations in the thiazide-sensitive sodium chloride cotransporter. Nat Genet 1996;12:24-30. [ Links ]
4. Jones AC, Chuck AJ, Arie EA, Green DJ, Doherty M. Diseases associated with calcium pyrophosphate deposition disease. Semin Arthritis Rheum 1992;22:188-202. [ Links ]
5. González Domínguez J, Escudero Contreras A, Pérez Guijo V, Martínez Sánchez FG, Caracuel Ruiz MA, Collantes Estévez E. Condrocalcinosis e hipomagnesemia: evolución clínicorradiológica. Reumatol Clin 2008;4:37-9. [ Links ]
6. Puchades MJ, González Rico MA, Pons S, Miguel A, Bonilla B. Alcalosis metabólica hipopotasémica: a propósito de un síndrome de Gitelman. Nefrología 2004;24,Suppl III:72-5. [ Links ]
7. Schlingmann KP, Konrad M, Seyberth HW. Genetics of hereditary disorders of magnesium homeostasis. Pediatr Nephrol 2004;19:13-25. [ Links ]
8. Martín V, Lafarga M, Garcia L, Rodrigo MD. Diagnóstico casual de un síndrome de Gitelman. Semergen 2014;40:95-8. [ Links ]
9. García Nieto V, Catambrana A, Müller D, Claverie-Martin F. Condrocalcinosis e Hipomagnesemia en un paciente portador del Gen cotransportador de una nueva mutación del CLNa sensible a tiazidas. Nefrología 2003;6:504-9. [ Links ]
10. Molina A, Mon C. Variabilidad Clínica del Síndrome de Gitelman. Nefrología 2006;26:504-6. [ Links ]
11. Richette P, Ayoub G, Lahalle S, Vicaut E, Badran AM, Joly F, et al. Hypomagnesemia assocaited with chondrocalcinosis: a cross-sectional study. Arhritis Rheum 2007;57:1496-501. [ Links ]
12. Caló L, Punzi L, Semplicini A. Hypomagnesemia and chondrocalcinosis in Bartter's and Gitelman's syndrome; review of the pathogenic mechanisms. Am J Nephrol 2000;20:347-50. [ Links ]

