










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
versión On-line ISSN 2173-2345versión impresa ISSN 1889-836X
Rev Osteoporos Metab Miner vol.8 no.4 Madrid oct./dic. 2016
REVISIÓN
El estrés oxidativo como posible diana terapéutica en la osteoporosis asociada al envejecimiento
Oxidative stress as a possible therapeutic target for osteoporosis associated with aging
Portal-Núñez S.1,4, de la Fuente M.2,4, Díez A.3,4 y Esbrit P.1,4
1 Área de Reumatología y Metabolismo Óseo - Instituto de Investigación Sanitaria-Fundación Jiménez Díaz - UAM - Madrid (España)
2 Departamento de Fisiología Animal II - Universidad Complutense - Madrid (España)
3 Hospital del Mar-IMIM-Universidad Autónoma de Barcelona - Barcelona (España)
4 Red Temática de Investigación Cooperativa en Envejecimiento y Fragilidad (RETICEF) - Instituto de Salud Carlos III - Madrid (España)
Este trabajo ha sido financiado con Ayudas de la Fundación Española de Investigación Ósea y del Metabolismo Mineral (Beca FEIOMM de Investigación Traslacional 2015), del Instituto de Salud Carlos III (RD12/0043/0022, PI11/00449, RD06/0013/1002, RD12/0043/0018 and RD12/ 0043/0008). SP-N disfruta un contrato RETICEF (RD06/0013/1002 y RD12/0043/0008).
Dirección para correspondencia
RESUMEN
La osteoporosis senil o involutiva es un problema de primera magnitud en el mundo desarrollado. Estudios recientes apuntan al aumento del estrés oxidativo asociado al envejecimiento -cronológico o biológico- como un factor importante en su desarrollo. En esta revisión nos centraremos en las alteraciones del tejido óseo con la edad, en el origen del estrés oxidativo y su influencia negativa en el tejido óseo. Finalmente, abordaremos las posibles terapias antiestrés oxidativo que actualmente se encuentran en desarrollo en esta patología.
Palabras clave: estrés oxidativo, osteoporosis, envejecimiento, fragilidad.
SUMMARY
Senile or involutional osteoporosis is a major problem in the developed world. Recent studies point to increased oxidative stress associated with aging, whether biological or chronological, as an important factor in its development. In this review paper, we focus on bone tissue disorders related to aging, the source of oxidative stress and negative influence on bone tissue. Finally, we consider the potential oxidative stress therapies currently being developed for this disease.
Key words: oxidative stress, osteoporosis, aging, fragility.
Introducción
El envejecimiento de la población de los países industrializados ha dado lugar al aumento de la prevalencia e incidencia de la osteoporosis. Se estima que aproximadamente 200 millones de personas en el mundo sufren esta patología1.
La osteoporosis, definida clásicamente como una disminución de la masa y la calidad ósea que incrementa el riesgo de fractura2, está muy relacionada con el envejecimiento, aunque los factores implicados no han sido totalmente identificados. Entre los factores relacionados con la osteoporosis involutiva se incluyen: el déficit estrogénico tras la menopausia3, el tratamiento con glucocorticoides4, la diabetes mellitus (DM), principalmente la tipo 25; la insuficiencia renal6 (causante de hiperparatiroidismo secundario); y, más recientemente, el aumento del estrés oxidativo, asociado a muchas de las situaciones anteriores7. En esta revisión profundizaremos en el papel del estrés oxidativo en el metabolismo óseo, así como sobre las posibles alternativas farmacológicas para paliar sus efectos deletéreos en la osteoporosis.
Alteraciones óseas asociadas al envejecimiento
El hueso es un tejido en continuo proceso de remodelado, con gran capacidad regenerativa y de adaptación a los cambios fisiológicos. Este proceso tiene lugar en las conocidas como unidades de remodelado óseo, constituidas por diferentes tipos celulares: osteoclastos, osteoblastos y osteocitos (osteoblastos completamente diferenciados embebidos en la matriz mineralizada y verdaderos orquestadores del proceso de remodelado)8. El remodelado óseo está altamente regulado por factores genéticos, mecánicos, hormonales y por factores locales del hueso, lo que determina el resultado del balance óseo.
El pico de masa ósea se alcanza durante la pubertad en mujeres y algo más tarde en varones. Estos alcanzan una masa ósea superior, presentando huesos más grandes y más anchos, mientras que en el caso de las mujeres son más pequeños y de menor diámetro y espesor cortical. A partir de la tercera década de la vida se observa en ambos sexos un balance óseo negativo (con predominio de la resorción ósea) que conlleva una pérdida paulatina de masa ósea similar en ambos sexos, inicialmente en el hueso trabecular y posteriormente en el cortical3. Este declive se acelera tras la menopausia en la mujer debido a la pérdida de estrógenos, agentes con probada capacidad antioxidante, lo que contribuye a mantener una masa ósea inferior al de los varones durante el envejecimiento. Con la edad, se producen alteraciones metabólicas que afectan al hueso: cambios neuromusculares relacionados con la falta de movilidad; aumento de la producción endógena de glucocorticoides; e insuficiencia renal, con una menor síntesis de calcitriol. Además, durante el envejecimiento las fibras de colágeno óseo sufren cambios estructurales y el hueso pierde la capacidad de reparar microfracturas9. Todo ello contribuye al aumento de incidencia de fracturas.
La mayoría de los conceptos actuales sobre el desarrollo de la osteoporosis senil se han obtenido a partir de estudios en modelos experimentales, fundamentalmente en roedores. Sin embargo, a la hora de interpretar estos resultados hay que tener en cuenta algunas particularidades óseas que presentan los roedores con respecto a los humanos, como son el continuo modelado óseo a partir de la placa de crecimiento, la ausencia de menopausia, así como la carencia de sistema haversiano en el hueso cortical. Sin embargo, al igual que en humanos, en roedores se ha demostrado una pérdida de masa ósea asociada a la edad10. Así, en ratas se ha observado un deterioro estructural y de la capacidad regenerativa de los huesos largos con la edad del animal11. La pérdida de masa ósea en ratas envejecidas tiene relación con una disminución de la maduración osteoblástica y con un aumento del número de osteoclastos frente al de osteoblastos en el hueso trabecular12. Así mismo, en ratones consanguíneos en los que la masa ósea está regulada principalmente por factores genéticos, la pérdida de masa ósea asociada a la edad puede llegar a suponer hasta el 10% de la masa ósea total, que se atribuye a un descenso del remodelado óseo13-16.
De modo similar a lo observado en roedores, en humanos predomina al principio la pérdida de hueso trabecular con la edad, sobre todo en las mujeres17, relacionada en parte con un descenso en la actividad física y, como consecuencia, de los estímulos mecánicos en el tejido óseo18. A partir de los 70 años cobra más protagonismo la disminución del espesor cortical y se produce un aumento concomitante de la porosidad intracortical del fémur, mientras aumenta el área medular, tanto en varones como en mujeres19. Estos cambios se relacionan con el incremento del riesgo de fracturas osteoporóticas. Sin embargo, tanto en ratones como en humanos las propiedades mecánicas del tejido óseo están relativamente conservadas gracias a un aumento mantenido de mineral subperióstico, lo que aumenta el momento de inercia20.
Mecanismos asociados al envejecimiento óseo
Los mecanismos moleculares subyacentes a la osteoporosis involutiva se han comenzado a dilucidar en los últimos años. Asociada a la edad, se ha observado una disminución del cociente osteoprotegerina (OPG)/ligando del receptor activador del factor nuclear (NF)-κB (RANKL), siendo dicho cociente un modulador importante del remodelado óseo21. Tanto OPG como RANKL son producidos y secretados al medio extracelular por las células osteoblásticas y osteocitos. De hecho, estudios en ratones indican que los osteocitos producen la mayoría del RANKL, influyendo así de manera directa en el remodelado óseo23,23. La OPG es un receptor señuelo soluble que capta RANKL en el medio extracelular (o en la superficie de los osteoblastos) y le impide unirse a su receptor (RANK) en las células de estirpe osteoclástica, impidiendo así la maduración y activación de los osteoclastos. Así, la relación OPG/RANKL es un factor importante en el balance anabólico/catabólico durante el remodelado óseo fisiológico24. De este modo, la disminución de la relación OPG/RANKL con la edad es compatible con el aumento de precursores osteoclásticos en la médula ósea de ratones viejos25. La apoptosis osteocítica juega un papel importante en la pérdida de masa ósea asociada a la edad y a la inmovilización o la falta de estímulos mecánicos26-28 y, además, se asocia a un incremento de la expresión de RANKL21. Por otra parte, en ratones viejos de la cepa C57BL/6 se ha observado un aumento de la producción de glucocorticoides endógenos -a través de la activación de la enzima 11β-hidroxiesteroide deshidrogenasa tipo 1-, un hecho relacionado con la reducción de viabilidad de las células óseas (osteoblastos y osteoclastos) y de la angiogénesis, un proceso fundamental en la formación ósea29.
Hay diversos factores que parecen afectar la tasa de reparación de fracturas con la edad30. Durante el envejecimiento existe una disminución de osteoprogenitores en la médula ósea, que ocurre paralelamente a un incremento de la adipogénesis31. Cabe señalar que tanto osteoblastos como adipocitos comparten una misma célula mesenquimal precursora diferenciable a uno u otro linaje en función del microambiente al que se ven expuestas estas células. Además, se ha demostrado que en los osteoblastos procedentes de ratones viejos la producción de RANKL aumenta en paralelo a la disminución de la de expresión de OPG y que esta alteración se traduce en una mayor osteoclastogénesis y actividad osteoclástica21,25. Es de interés resaltar que, también asociada a la edad, se produce una disminución del número de células endoteliales y de la angiogénesis, que puede contribuir negativamente al proceso de reparación ósea en sujetos de edad avanzada32.
Recientemente se ha observado un aumento de masa ósea y una disminución del riesgo de fracturas en sujetos ancianos tratados con antagonistas de los receptores de angiotensina II33. El aparente efecto beneficioso para el hueso de estos fármacos se atribuye a la acción inhibitoria de la angiotensina II sobre diversos marcadores de diferenciación osteoblástica, como el factor de transcripción relacionado con runt 2 (Runx2), indispensable para la diferenciación osteoblástica, la osteocalcina y la fosfatasa alcalina34, y al incremento del RANKL, que favorece la diferenciación osteoclástica35. Estos datos sugieren que la hipertensión arterial prevalente en los ancianos podría también contribuir a la osteoporosis involutiva.
El producto del gen Sost, específico de los osteocitos y conocido como esclerostina, es un potente inhibidor de la formación ósea a través de su unión a los receptores relacionados con la lipoproteína de baja densidad 5 y 6, inhibiendo así la vía canónica de Wnt. Estudios recientes han demostrado que la esclerostina circulante aumenta en mujeres post-menopaúsicas y con la edad en ambos sexos, lo que podría tener una influencia negativa sobre la masa ósea36,37.
Actualmente se sabe que un importante modulador del envejecimiento celular es el producto del gen Klotho38, una proteína transmembrana que actúa como co-receptor del factor de crecimiento fibroblástico (FGF) 23 producido por los osteocitos e inductor de fosfaturia. Ratones deficientes en el gen Klotho sufren un envejecimiento acelerado y osteopenia, caracterizada por una reducción (20-40%) del grosor cortical en fémur, tibia y vértebras, y un bajo remodelado óseo con un descenso muy acusado de la formación ósea cortical. Las células estromales de la médula ósea de estos ratones presentan una disminución de su capacidad de formación de nódulos mineralizados y de la actividad de fosfatasa alcalina39. Paradójicamente, estos ratones deficientes en Klotho poseen un aumento de hueso trabecular en las vértebras y las metáfisis de los huesos largos; un efecto que los autores atribuyen a una activación selectiva de la vía Wnt sobre el componente trabecular. Klotho interacciona con la vía Wnt a través de su producto secretado, el cual se une a ligandos de esta vía inhibiendo su acción, de ahí que la ausencia de Klotho pueda dar lugar a la activación de la vía Wnt35. Por otro lado, se ha demostrado que ratones con ausencia de telomerasa presentan un aumento de la senescencia celular, así como un descenso en la masa ósea a los 3 meses del nacimiento, asociados a una reducción de formación ósea y de osteoblastogénesis40. Al parecer, esta reducción se debe a que los ratones sin telomerasa presentan osteoblastos poco diferenciados y al ambiente proinflamatorio que promueve la actividad osteoclástica.
El estrés oxidativo como factor patogénico en la osteoporosis involutiva
El envejecimiento se puede considerar como una consecuencia del desequilibrio entre los agentes oxidantes producidos naturalmente en el metabolismo celular y las defensas antioxidantes, con predominio de los primeros. Esto se conoce como estrés oxidativo, el cual conlleva la oxidación de biomoléculas y la pérdida funcional de las células41,42. El aumento de estrés oxidativo, que se realiza fundamentalmente en la mitocondria, tiene como base el exceso de producción de especies reactivas de oxígeno (ERO), como el anión superóxido (O2.-), los radicales hidroxilo (.OH) y el peróxido de hidrógeno (H2O2).
Este aumento no puede ser adecuadamente equilibrado por los sistemas antioxidantes, como la enzima superóxido dismutasa (SOD), la catalasa (CAT), las enzimas del ciclo del glutatión (glutatión peroxidasa y glutatión reductasa) y la tiorredoxina, entre otros. El exceso de ERO con la edad cronológica (y/o biológica), produce la oxidación de ADN, proteínas y lípidos e induce la fosforilación de la proteína mitocondrial p66shc, lo que lleva a la muerte celular7,43-45 (Figura 1). Recientemente, se ha comprobado que el estrés oxidativo tiene también importantes funciones en la señalización celular46,47, y en este contexto, los ERO se pueden considerar segundos mensajeros de la respuesta inflamatoria. De hecho, oxidación e inflamación son dos procesos íntimamente relacionados que aumentan con la edad48.

Aunque algunos investigadores han planteado dudas sobre si el estrés oxidativo es causa o consecuencia del envejecimiento, en los últimos años se ha implicado al mismo en el deterioro óseo49. En este sentido, utilizando diversos modelos animales: con envejecimiento prematuro, osteoporosis por déficit estrogénico (tras ovariectomía), o diabéticos, se constata un aumento de marcadores de estrés oxidativo en relación con una disminución de la formación ósea50-54.Los mecanismos por los que el estrés oxidativo induce efectos deletéreos en el tejido óseo no son aun bien conocidos. El aumento de ERO conduce a la estabilización de factores de transcripción forkhead box O (FoxO), una importante familia de factores de transcripción reguladores de multitud de genes con funciones tales como el control del metabolismo de la glucosa, la génesis tumoral y la defensa celular contra el estrés oxidativo55. FoxO 1 y 3 se expresan en hueso56, donde parecen jugar un papel clave en el mantenimiento de la formación ósea56. Se ha demostrado que la eliminación genética de FoxOs en ratones incrementa el estrés oxidativo en el hueso e induce la pérdida de masa ósea trabecular y cortical, asociada al aumento de apoptosis osteoblástica/osteocítica y a una disminución de formación ósea57. La activación de FoxO por fosforilación conlleva su acoplamiento con la β-catenina57, provocando la inducción de genes de respuesta al estrés oxidativo, como GADD45 y CAT58. De hecho, la acción protectora del estrés oxidativo de la proteína Klotho citada anteriormente parece mediada por la activación de FoxOs39. Por otro lado, la activación de FoxO previene que la β-catenina actúe como factor de transcripción para estimular la proliferación y diferenciación de los osteoblastos56.
El aumento de ERO en las células óseas produce daños en el ADN genómico y apoptosis de los osteoblastos y osteocitos. Además, la peroxidación lipídica dependiente de lipoxigenasas activadas por el estrés oxidativo juega un papel importante en la pérdida ósea asociada al envejecimiento. Esto se evidencia al analizar la expresión de las lipoxigenasas Alox12 y Alox15 y la formación del 4-hidroxinonenal, un producto de peroxidación lipídica, aumentadas en el hueso de ratones viejos59. Además, se ha demostrado que los productos de oxidación lipídica inhiben la acción de factores osteogénicos60.
Por otro lado, el aumento de ERO ha sido relacionado con un incremento de la osteoclastogénesis y de la actividad de los osteoclastos61,62. Recientemente se ha demostrado que la enzima nicotinamida adenina dinucleótido fosfato oxidasa 4 (NOX 4) juega un papel fundamental en la osteoclastogénesis. Ratones deficientes para esta enzima, que produce constitutivamente ERO, tienen una elevada masa ósea y un déficit de los marcadores osteoclásticos; además, en muestras óseas humanas la actividad osteoclástica elevada se correlacionada con un aumento de actividad de NOX 463. Por otro lado, cabe señalar que en situaciones de aumento de ERO asociado a la DM experimental, existen resultados dispares. Mientras que algunos autores han observado un aumento de la actividad osteoclástica64, se ha sugerido que podría estar en relación con la mayor severidad de la DM65, sin embargo, en otros modelos de DM se observa una actividad osteoclástica disminuida66. De hecho, estudios utilizando pre-osteoclastos murinos incubados en presencia de alta glucosa parecen confirmar su efecto inhibitorio sobre los osteoclastos67. Así pues, diferencias en el grado de la DM, la cepa y la edad del animal, podrían contribuir al diferente estado de la resorción ósea observado en diferentes modelos diabéticos65,68.
Posibles terapias antiestrés oxidativo en la osteoporosis senil
El desarrollo de nuevas terapias anabólicas para la osteoporosis que combinen el aumento de la masa ósea con su capacidad para neutralizar los efectos perniciosos del estrés oxidativo es de sumo interés. Una aproximación intuitiva para evitar el deterioro óseo con la edad estaría basada en la administración de agentes antioxidantes. Sin embargo, se ha apuntado que los antioxidantes clásicos, como puede ser la CAT o la N-acetilcisteína, ejercerían efectos indeseados en el tejido óseo, ya que actuarían como auténticos agentes antiosteoclastogénicos interfiriendo con el remodelado óseo69. Además, este tipo de agentes inhiben la vía canónica de Wnt/β-catenina cuya activación es de suma importancia para el mantenimiento de la formación ósea, en parte induciendo el secuestro de la proteína activadora dishevelled por la proteína reguladora del equilibrio redox, nucleoredoxina70.
Recientemente, se ha relacionado el efecto anabólico óseo asociado a la administración intermitente de la parathormona (PTH) con sus propiedades antiestrés oxidativo, como son el descenso de la cantidad de ERO, la inhibición de la fosforilación de p66shc y el aumento de la cantidad de glutatión total69. La ventaja de este tratamiento con la PTH frente a los antioxidantes clásicos la determina su acción estimuladora del remodelado óseo, con predominio de la formación ósea en parte a través de su interacción con la vía de Wnt/β-catenina (Figura 2). En este contexto, se ha demostrado in vitro que los fragmentos N-terminal (1-36) (homólogo con la PTH) y C-terminal (107-109) de la proteína relacionada con la PTH (PTHrP), son capaces de contrarrestar el estrés oxidativo inducido por H2O2 en células osteoprogenitoras en relación con su acción osteogénica52,71.

Estudios in vitro y en modelos animales sugieren que el resveratrol, un compuesto bifenólico del grupo de los antioxidantes polifenólicos, presente en la piel de la uva y de otros frutos72,73, podría ser un potencial agente antiosteoporótico. Este compuesto aumenta la proliferación y diferenciación de pre-osteoblastos de ratón MC3T3-E1 in vitro73. Además, se ha demostrado que la administración de resveratrol a células mesenquimales derivadas de células madre embrionarias humanas induce la expresión de Runx274 y su diferenciación a osteoblastos maduros75. Este mecanismo de acción del resveratrol parece estar mediado por la activación de la deacetilasa Sirt1 que aumenta la expresión de FoxO3a y la formación de un complejo con el resveratrol, incrementando así la expresión de Runx2 (Figura 3). Sirt1 podría también aumentar la actividad de Runx2 directamente al deacetilar a este factor de transcripción en células pre-osteoblásticas. En un reciente trabajo en ratas viejas se ha demostrado que la administración de resveratrol (10 mg/kg diariamente durante 10 semanas) mejora la calidad ósea y las propiedades biomecánicas del hueso osteoporótico76. Aunque estos resultados pre-clínicos son prometedores, todavía no hay datos contrastados que confirmen la eficacia del resveratrol en la osteoporosis senil en humanos. Sin embargo, cabe destacar un reciente estudio llevado a cabo en pacientes obesos y osteopénicos, en los que la administración oral de resveratrol (1 g diario durante 16 semanas) incrementó la masa ósea de manera significativa, así como la cantidad de fosfatasa alcalina ósea, frente al grupo placebo77. Recientemente se ha publicado que ratones deficientes en Sirt6, otra deacetilasa relacionada con la respuesta al estrés oxidativo, presentan un fenotipo osteoporótico a edades tempranas. La ausencia de Sirt6 se asocia a la sobreexpresión de Runx2, osterix y OPG, así como al aumento del inhibidor de la vía Wnt, Dickkopf 1, que conduce a un déficit de maduración osteoblástica y osteoclástica78. Estos datos apuntan a que Sirt6 podría ser una diana terapéutica en la osteoporosis involutiva.

Por otro lado, el exceso de glucocorticoides también induce estrés oxidativo. En esta situación, el estrés oxidativo observado en el retículo plasmático puede ser revertido por la fosforilación del factor de iniciación de la traducción 2α, que interrumpe la traducción proteica. Recientemente se ha demostrado que el salubrinal, un compuesto que evita esta defosforilación, evita el déficit de mineralización de los osteoblastos tratados con glucocorticoides in vitro, así como la apoptosis osteoblástica y osteocítica en un modelo murino de osteoporosis por administración de prednisolona79.
Conclusiones
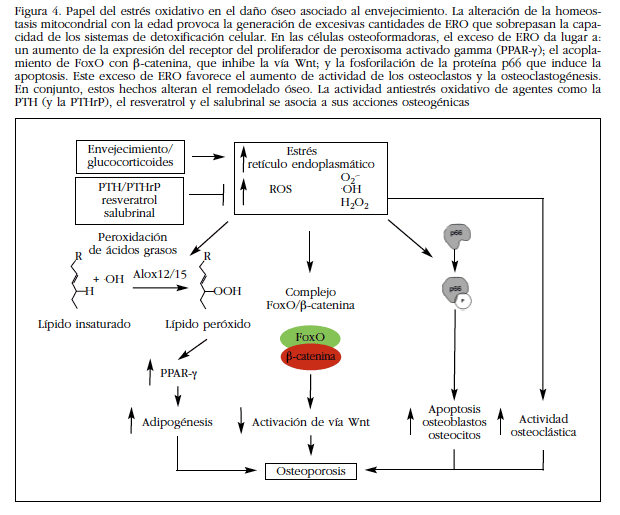
El progresivo envejecimiento de la población en el mundo desarrollado conlleva un aumento de las patologías musculoesqueléticas, que incluyen la osteoporosis. La osteoporosis y el aumento de fragilidad de la población senil constituyen un reto socio-económico de primera magnitud. Diferentes factores contribuyen al deterioro óseo en los ancianos, entre los que destaca como elemento común el incremento de estrés oxidativo (Figura 4). Así pues, reducir el estrés oxidativo podría ser una herramienta útil para combatir la osteoporosis involutiva. Sin embargo, el hecho de que los compuestos antiestrés oxidativo podrían interferir con el remodelado óseo o con vías anabólicas claves para la formación ósea, como la vía Wnt, requiere ciertas consideraciones previas a su uso terapéutico. Hay que tener en cuenta también el papel fisiológico de las ERO, que actúan como mensajeros secundarios de muchas vías metabólicas; por tanto, su inhibición no controlada podría dar lugar a efectos secundarios no deseados en las células óseas. Así pues, son necesarias nuevas investigaciones que determinen el verdadero efecto de las terapias antioxidantes y sus pautas adecuadas de administración, evitando acciones deletéreas sobre el remodelado óseo. Teniendo en cuenta estas consideraciones, las terapias enfocadas a neutralizar el estrés oxidativo para prevenir o alterar el curso de la osteoporosis involutiva supondrían un avance sanitario evidente.
Conflicto de intereses
Los autores declaran no tener conflictos de intereses.
Bibliografía
1. Cooper C, Campion G, Melton LJ. Hip fractures in the elderly: a world-wide projection. Osteoporos Int. 1992;2:285-9. [ Links ]
2. Reginster J-Y, Burlet N. Osteoporosis: a still increasing prevalence. Bone. 2006;38:S4-9. [ Links ]
3. Khosla S, Riggs BL. Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin North Am. 2005;34:1015-30, xi. [ Links ]
4. Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 2003;48:3224-9. [ Links ]
5. Vestergaard P, Rejnmark L, Mosekilde L. Diabetes and its complications and their relationship with risk of fractures in type 1 and 2 diabetes. Calcif Tissue Int. 2009;84:45-55. [ Links ]
6. Miller PD. Bone disease in CKD: a focus on osteoporosis diagnosis and management. Am J Kidney Dis. 2014;64:290-304. [ Links ]
7. Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266-300. [ Links ]
8. Eriksen EF. Cellular mechanisms of bone remodeling. Rev. Endocr. Metab Disord. 2010;11:219-27. [ Links ]
9. Bailey AJ, Knott L. Molecular changes in bone collagen in osteoporosis and osteoarthritis in the elderly. Exp Gerontol. 1999;34:337-51. [ Links ]
10. Wang L, Banu J, McMahan CA, Kalu DN. Male rodent model of age-related bone loss in men. Bone. 2001;29:141-8. [ Links ]
11. Liang CT, Barnes J, Seedor JG, Quartuccio HA, Bolander M, Jeffrey JJ, et al. Impaired bone activity in aged rats: alterations at the cellular and molecular levels. Bone. 1992;13:435-41. [ Links ]
12. Roholl PJ, Blauw E, Zurcher C, Dormans JA, Theuns HM. Evidence for a diminished maturation of preosteoblasts into osteoblasts during aging in rats: an ultrastructural analysis. J Bone Miner Res. 1994;9:355-66. [ Links ]
13. Kobayashi Y, Goto S, Tanno T, Yamazaki M, Moriya H. Regional variations in the progression of bone loss in two different mouse osteopenia models. Calcif Tissue Int. 1998;62:426-36. [ Links ]
14. Ferguson VL, Ayers RA, Bateman TA, Simske SJ. Bone development and age-related bone loss in male C57BL/6J mice. Bone. 2003;33:387-98. [ Links ]
15. Turner CH, Hsieh Y-F, Müller R, Bouxsein ML, Baylink DJ, Rosen CJ, et al. Genetic Regulation of Cortical and Trabecular Bone Strength and Microstructure in Inbred Strains of Mice. J Bone Miner Res. 2000;15:1126-31. [ Links ]
16. Weiss A, Arbell I, Steinhagen-Thiessen E, Silbermann M. Structural changes in aging bone: osteopenia in the proximal femurs of female mice. Bone. 1991;12:165-72. [ Links ]
17. Schaadt O, Bohr H. Different trends of age-related diminution of bone mineral content in the lumbar spine, femoral neck, and femoral shaft in women. Calcif Tissue Int. 1988;42:71-6. [ Links ]
18. Hamrick MW, Ding K-H, Pennington C, Chao YJ, Wu Y-D, Howard B, et al. Age-related loss of muscle mass and bone strength in mice is associated with a decline in physical activity and serum leptin. Bone. 2006;39:845-53. [ Links ]
19. Feik SA, Thomas CD, Clement JG. Age-related changes in cortical porosity of the midshaft of the human femur. J Anat. 1997;191:407-16. [ Links ]
20. Stein MS, Thomas CD, Feik SA, Wark JD, Clement JG. Bone size and mechanics at the femoral diaphysis across age and sex. J Biomech. 1998;31:1101-10. [ Links ]
21. Cao J, Venton L, Sakata T, Halloran BP. Expression of RANKL and OPG Correlates With Age-Related Bone Loss in Male C57BL/6 Mice. J Bone Miner Res. 2003;18:270-7. [ Links ]
22. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231-4. [ Links ]
23. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235-41. [ Links ]
24. Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev. 2008;29:155-92. [ Links ]
25. Cao JJ, Wronski TJ, Iwaniec U, Phleger L, Kurimoto P, Boudignon B, et al. Aging Increases Stromal/ Osteoblastic Cell-Induced Osteoclastogenesis and Alters the Osteoclast Precursor Pool in the Mouse. J Bone Miner Res. 2005;20:1659-68. [ Links ]
26. Jilka RL, O'Brien CA. The Role of Osteocytes in Age-Related Bone Loss. Curr Osteoporos Rep. 2016;14:16-25. [ Links ]
27. Jilka RL, Noble B, Weinstein RS. Osteocyte apoptosis. Bone. 2013;54:264-71. [ Links ]
28. Bikle DD, Sakata T, Halloran BP. The impact of skeletal unloading on bone formation. Gravit Space Biol Bull. 2003;16:45-54. [ Links ]
29. Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O'Brien CA, et al. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell. 2010;9:147-61. [ Links ]
30. Gruber R, Koch H, Doll BA, Tegtmeier F, Einhorn TA, Hollinger JO. Fracture healing in the elderly patient. Exp Gerontol. 2006;41:1080-93. [ Links ]
31. Gimble JM, Zvonic S, Floyd ZE, Kassem M, Nuttall ME. Playing with bone and fat. J Cell Biochem. 2006;98:251-66. [ Links ]
32. Edelberg JM, Reed MJ. Aging and angiogenesis. Front. Biosci. 2003;8:s1199-209. [ Links ]
33. Rejnmark L, Vestergaard P, Mosekilde L. Treatment with beta-blockers, ACE inhibitors, and calcium-channel blockers is associated with a reduced fracture risk: a nationwide case-control study. J Hypertens. 2006;24:581-9. [ Links ]
34. Franceschi RT. The developmental control of osteoblast-specific gene expression: role of specific transcription factors and the extracellular matrix environment. Crit Rev Oral Biol Med. 1999;10:40-57. [ Links ]
35. Shimizu H, Nakagami H, Osako MK, Hanayama R, Kunugiza Y, Kizawa T, et al. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008;22:2465-75. [ Links ]
36. Ardawi M-SM, Al-Kadi HA, Rouzi AA, Qari MH. Determinants of serum sclerostin in healthy pre-and postmenopausal women. J Bone Miner Res. 2011;26:2812-22. [ Links ]
37. Mödder UI, Hoey KA, Amin S, McCready LK, Achenbach SJ, Riggs BL, et al. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. J Bone Miner Res. 2011;26:373-9. [ Links ]
38. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45-51. [ Links ]
39. Kuro-o M. Klotho and aging. Biochim. Biophys. Acta. 2009;1790:1049-58. [ Links ]
40. Saeed H, Abdallah BM, Ditzel N, Catala-Lehnen P, Qiu W, Amling M, et al. Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J Bone Miner Res. 2011;26:1494-505. [ Links ]
41. Harman D. About "Origin and evolution of the free radical theory of aging: a brief personal history, 1954-2009". Biogerontology. 2009;10:783. [ Links ]
42. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298-300. [ Links ]
43. De la Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des. 2009;15:3003-26. [ Links ]
44. Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J. Biol Chem. 2009;284:27438-48. [ Links ]
45. Almeida M, Han L, Martin-Millan M, O'Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor-to forkhead box O-mediated transcription. J Biol Chem. 2007;282:27298-305. [ Links ]
46. Bindoli A, Rigobello MP. Principles in redox signaling: from chemistry to functional significance. Antioxid Redox Signal. 2013;18:1557-93. [ Links ]
47. Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact. 2014;224:164-75. [ Links ]
48. Vida C, González EM, De la Fuente M. Increase of oxidation and inflammation in nervous and immune systems with aging and anxiety. Curr Pharm Des. 2014;20:4656-78. [ Links ]
49. Hamada Y, Kitazawa S, Kitazawa R, Fujii H, Kasuga M, Fukagawa M. Histomorphometric analysis of diabetic osteopenia in streptozotocin-induced diabetic mice: a possible role of oxidative stress. Bone. 2007;40:1408-14. [ Links ]
50. Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282:27285-97. [ Links ]
51. Brunet A. (Aging and the control of the insulin-FOXO signaling pathway). Médecine Sci. M/S. 2012;28:316-20. [ Links ]
52. de Castro LF, Lozano D, Portal-Núñez S, Maycas M, De la Fuente M, Caeiro JR, et al. Comparison of the skeletal effects induced by daily administration of PTHrP (1-36) and PTHrP (107-139) to ovariectomized mice. J Cell Physiol. 2012;227:1752-60. [ Links ]
53. Portal-Núñez S, Manassra R, Lozano D, Acitores A, Mulero F, Villanueva-Peñacarrillo ML, et al. Characterization of skeletal alterations in a model of prematurely aging mice. Age (Dordr). 2013;35:383-93. [ Links ]
54. Portal-Núñez S, Cruces J, Gutiérrez-Rojas I, Lozano D, Ardura JA, Villanueva-Peñacarrillo ML, et al. The vertebrae of prematurely aging mice as a skeletal model of involutional osteoporosis. Histol Histopathol. 2013;28:1473-81. [ Links ]
55. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410-25. [ Links ]
56. Ambrogini E, Almeida M, Martin-Millan M, Paik J-H, Depinho RA, Han L, et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010;11:136-46. [ Links ]
57. Essers MAG, de Vries-Smits LMM, Barker N, Polderman PE, Burgering BMT, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181-4. [ Links ]
58. Katoh M, Katoh M. Human FOX gene family (Review). Int J Oncol. 2004;25:1495-500. [ Links ]
59. Huang MS, Morony S, Lu J, Zhang Z, Bezouglaia O, Tseng W, et al. Atherogenic phospholipids attenuate osteogenic signaling by BMP-2 and parathyroid hormone in osteoblasts. J Biol Chem. 2007;282:21237-43. [ Links ]
60. Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, et al. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112:915-23. [ Links ]
61. Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest. 1990;85:632-9. [ Links ]
62. Lee NK, Choi YG, Baik JY, Han SY, Jeong D-W, Bae YS, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852-9. [ Links ]
63. Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, et al. NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Invest. 2013;123:4731-8. [ Links ]
64. Botolin S, Faugere M-C, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146:3622-31. [ Links ]
65. Motyl K, McCabe LR. Streptozotocin, type I diabetes severity and bone. Biol Proced Online. 2009;11:296-315. [ Links ]
66. Verhaeghe J, Thomsen JS, van Bree R, van Herck E, Bouillon R, Mosekilde L. Effects of exercise and disuse on bone remodeling, bone mass, and biomechanical competence in spontaneously diabetic female rats. Bone. 2000;27:249-56. [ Links ]
67. Wittrant Y, Gorin Y, Woodruff K, Horn D, Abboud HE, Mohan S, et al. High d(+)glucose concentration inhibits RANKL-induced osteoclastogenesis. Bone. 2008;42:1122-30. [ Links ]
68. Portal-Núñez S, Ardura JA, Lozano D, Bolívar OH, López-Herradón A, Gutiérrez-Rojas I, et al. Adverse effects of diabetes mellitus on the skeleton of aging mice. J Gerontol A Biol Sci Med. Sci. 2016;71:290-9. [ Links ]
69. Jilka RL, Almeida M, Ambrogini E, Han L, Roberson PK, Weinstein RS, et al. Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell. 2010;9:851-67. [ Links ]
70. Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. 2006;8:501-8. [ Links ]
71. Lozano D, Fernández-de-Castro L, Portal-Núñez S, López-Herradón A, Dapía S, Gómez-Barrena E, et al. The C-terminal fragment of parathyroid hormone-related peptide promotes bone formation in diabetic mice with low-turnover osteopaenia. Br J Pharmacol. 2011;162:1424-38. [ Links ]
72. Farghali H, Kutinová Canová N, Lekiæ N. Resveratrol and related compounds as antioxidants with an allosteric mechanism of action in epigenetic drug targets. Physiol Res. 2013;62:1-13. [ Links ]
73. Mizutani K, Ikeda K, Kawai Y, Yamori Y. Resveratrol stimulates the proliferation and differentiation of osteoblastic MC3T3-E1 cells. Biochem Biophys Res Commun. 1998;253:859-63. [ Links ]
74. Tseng P-C, Hou S-M, Chen R-J, Peng H-W, Hsieh C-F, Kuo M-L, et al Resveratrol promotes osteogenesis of human mesenchymal stem cells by upregulating RUNX2 gene expression via the SIRT1/FOXO3A axis. J Bone Miner Res. 2011;26:2552-63. [ Links ]
75. Shakibaei M, Shayan P, Busch F, Aldinger C, Buhrmann C, Lueders C, et al. Resveratrol mediated modulation of Sirt-1/Runx2 promotes osteogenic differentiation of mesenchymal stem cells: potential role of Runx2 deacetylation. PLoS One. 2012;7:e35712. [ Links ]
76. Tresguerres IF, Tamimi F, Eimar H, Barralet J, Torres J, Blanco L, et al. Resveratrol as anti-aging therapy for agerelated bone loss. Rejuvenation Res. 2014;17:439-45. [ Links ]
77. Ornstrup MJ, Harsløf T, Kjær TN, Langdahl BL, Pedersen SB. Resveratrol increases bone mineral density and bone alkaline phosphatase in obese men: a randomized placebo-controlled trial. J Clin Endocrinol Metab. 2014;99:4720-9. [ Links ]
78. Sugatani T, Agapova O, Malluche HH, Hruska KA. SIRT6 deficiency culminates in low-turnover osteopenia. Bone. 2015;81:168-77. [ Links ]
79. Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone. 2015;73:60-8. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Sergio Portal Núñez
Área de Reumatología y Metabolismo Óseo
Instituto de Investigación Sanitaria-Fundación Jiménez Díaz
Avda. Reyes Católicos, 2
28040 Madrid (España)
Correo electrónico: sportal@fjd.es
Fecha de recepción: 19/02/2016
Fecha de aceptación: 13/06/2016
