










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkArs Pharmaceutica (Internet)
On-line version ISSN 2340-9894
Ars Pharm vol.57 n.4 Granada Oct./Dec. 2016
https://dx.doi.org/10.30827/ars.v57i4.5560
ORIGINAL ARTICLE
Synthesis and cytotoxic activity of tri-acyl ester derivatives of uridine in breast cancer cells
Síntesis y actividad citotóxica de derivados triesterificados de la uridina en células de cáncer de mama
Jhon Fernando Berrío Escobar1, Víctor Hugo Arango Carmona1, Elkin Galeano Jaramillo1, Diana Margarita Márquez Fernández1, María Elena Márquez Fernández2, Mauricio Camargo Guerrero3 y Alejandro Martínez Martínez1
1. Grupo Productos Naturales Marinos, Facultad de Ciencias Farmacéuticas y Alimentarias, Universidad de Antioquia UdeA, Calle 70 No 52-21, Medellín-Colombia.
2. Universidad Nacional Sede Medellín-Colombia. Facultad de Ciencias. Grupo Biotecnología Animal.
3. Grupo de Genética, Regeneración y Cáncer, Sede de Investigación Universitaria, Universidad de Antioquia, Medellín-Antioquia, Colombia.
Authors thank financing to the University of Antioquia (Project CODI CIQF-155 2012-2014, Strategy of Sustainability 2014-2015, Student instructor scholarship and Young ResearchersProgram) and Colciencias (Young Researchers Program)
SUMMARY
Aims: Synthesize tri-acyl ester derivatives of uridine, and evaluate its cytotoxicity against breast cancer cells line.
Methods: The tri-esterified uridine derivatives were obtained through Steglich esterification reaction by fatty and aromatic acids, and with acetic anhydride. An acetonide derivative from uridine was prepared with acid catalysis. Compounds were characterized by NMR spectroscopy (1H NMR and 13C NMR), and mass spectrometry. Derivatives were assessed in chinese hamster ovary (CHO-K1) and human breast cancer (MCF-7) cell lines.
Results: Five tri-acyl ester derivatives of uridine were obtained one acetic acid, three fatty acids (myristic acid, stearic acid and oleic acid) with an aromatic acid. The uridine per-acetylated and uridine acetonide were obtained in high yields, however, the tri-acyl ester derivatives of uridine with fatty and aromatic acids were obtained in moderate and low yields, respectively. The acetonide and compounds 2 and 3 exhibited a cell viability inhibition significant on both cell lines to the higher concentration.
Conclusions: Esterification method with coupling agents allowed obtained tri-acyl ester uridine derivatives with aliphatic and aromatic acids. However, significant cytotoxic activity (p<0.05) for uridine and its derivatives was not observed.
Key words: breast cancer; cytotoxic activity; esters synthesis; nucleosides; uridine derivatives.
RESUMEN
Objetivos: Sintetizar derivados triesterificados de la uridina y evaluar su citotóxicidad sobre una línea celular de cáncer de mama.
Métodos: Se prepararon derivados triesterificados de la uridina mediante la esterificación de Steglich para los ácidos grasos y aromáticos, y con anhídrido acético. Además se preparó el derivado acetonido mediante catálisis ácida. Los compuestos se caracterizaron por espectroscopia de RMN (RMN 1H y RMN 13C), y espectrometría de masas. Los derivados se evaluaron sobre líneas celulares de tumor de ovario de hámster chino (CHO) y de cáncer de mamá (MCF-7).
Resultados: Se obtuvieron cinco derivados triesterificados de la uridina, uno con ácido acético, tres con ácidos grasos (ácido mirístico, ácido esteárico y ácido oleico) y uno con ácido aromático. Los derivados de uridina per-acetilada y acetonido se obtuvieron con rendimientos altos, sin embargo los derivados con ácidos grasos y aromático, se obtuvieron con rendimientos moderados y bajo, respectivamente. El acetonido y los compuestos 2 y 3, exhibieron inhibición significativa de la viabilidad celular sobre ambas líneas a la concentración más alta evaluada.
Conclusiones: El método de esterificación con agentes de acoplamiento utilizado, permitió obtener derivados triesterificados de la uridina con ácidos grasos y aromáticos. No se observó actividad citotóxica significativa (p<0,05) para la uridina y sus derivados.
Palabras claves: actividad citotóxica; cáncer de mama; derivados de uridina; nucleósidos; síntesis de ésteres.
Introduction
Breast cancer is among the ten cancer types with higher world mortality1,2. Nucleoside analog drugs are widely used in cancer chemotherapy against soft tumors (leukemia and lynphome non-Hodking) and solid tumors (lung, liver, breast,)3,4. They show multiple action mechanisms: Inhibition of DNA synthesis, transcription and replication, DNA demethylation, destabilization of mitochondrial membrane and, activation of factors and apoptosis routes4-6. However, the anticancer nucleoside drugs presents pharmacokinetic (low selectivity by cancer cell and great chemical lability, and biological, among others) and pharmacodynamic (low incorporation in cells and intracellular inactivation and degradation by nucleoside deaminase and ectonucleotidase enzymes, respectively, among others) disadvantages3,7-9.
Investigations found that uridine (a natural and necessary nucleoside) and some her phosphorylated and glycosylated derivatives, exhibit cellular regulatory functions: membrane synthesis modulation, glycosyl-transferase cofactors activation/deactivation, purinergic receptor agonists P2Xn and P2Ym, UTP does ATP functions, when, low amounts of ATP is available, and some uridine derivatives show inhibition of DNA polymerases. Furthermore, the uridine and some biological derivatives behave as antimetabolites, when their extracellular concentrations is higher than those physiological level2,5,10-12.
Investigations have focused on the synthesis of uridine derivatives with best physical-chemical and biological properties, such as: synthesis of glycosilated derivatives of uridine with potential glycosil-transferase inhibitory activity11,12, Synthesis and citotoxic activity evaluation of uridine derivatives with aliphatic groups, 3'-C-ethynyluridine and uridine acetonide conjugates with triterpenoids, in breast cancer13,14, the triacetil-ester derivative of uridine has shown modulators effects in patients on the fluorouracil toxicity, exhibited when was used in high doses or longer treatments15.
Owing to the disadvantages and side effects by nucleosides previously displayed, it was proposed, synthesize uridine derivatives of ester type with different aliphatic and aromatic acids, and cell viability evaluation in breast cancer cell.
Materials and methods
Chemical
Reagent grade solvents: hexane and ethyl acetate 99% purity (Honeywell), dichloromethane, chloroform, acetone and methanol, N,N-dimethylformamide (DMF) 99% purity (Merck), and triethylamine (TEA) 99.9% (J.T. Baker). Uridine 99% (Alfaesar), acetic anhydride 99% (Merck), coupling agent: N,N'-diciclohexil-carbodiimide (DCC) 99%, and nucleophilic catalyst: 4-N,N-dimethylamino-pyridine (DMAP) 99% (Alfaesar), fatty acids (myristic, stearic and oleic acids) 99% (Merck), and aromatic acids (o-N-acetylanthranilic, p-nitrobenzoic and 3,5-dinitrobenzoic acids) 99% (Merck). Chromatography Column (silica gel 60N) with gradient: hexane/ethyl acetate and ethyl acetate/acetone mixes, and preparative Thin-Layer Chromatography with ethyl acetate/acetone mix as eluent. All compounds were characterized by nuclear magnetic resonance spectroscopy (1H-NMR, 13C-NMR, COSY H-H, HSQC and HMBC) on a Bruker NMR spectrometer (300 MHz, software Bruker TopSpin 3.1). Tetramethylsilane (TMS) was used as an internal standard, and deuterated chloroform (CDCl3) and deuterated dimethyl sulfoxide (DMSO-d6) 99% deuterium purity (Merck) were used as solvents. Mass spectra analyses were performed on an Agilent 6100 Series-LC/MS Equipment (electrospray ionization (ESI) and single quadrupole analyzer in positive mode) and a Agilent 6300-LC/MS Equipment (electrospray ionization (ESI) and Ion-Trap analyzer in positive mode) by direct injection method.
3',4',6'-O-triacetyl-uridine (1)
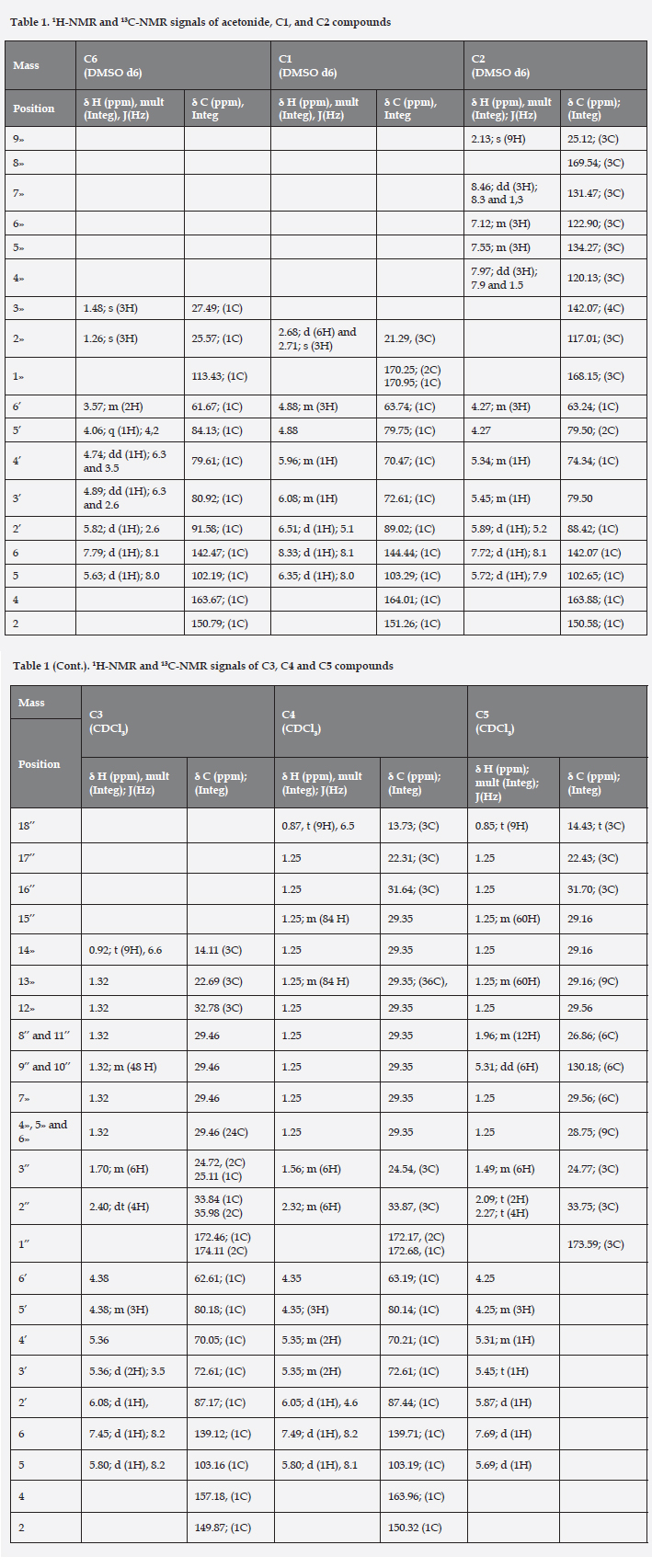
DMAP (5 mol-%) was aggregated to uridine (300 mg) solution in acetic anhydride/DMF/TEA (1:1:1) mixture. The reaction mixture was stirred at room temperature for 6 hours (for chemical reaction see figure 1) 16. The product was isolated as yellow solid (quantitative yield), Melting point: 127-128 oC. 1H NMR and 13C NMR data are in table 1. ESI-MS (positive mode) m/z: Calculated for C15H18N2O9: 370.3114; found, 393.1 [M+Na]+.

General procedure for the synthesis of tri-acyl ester derivatives
The esterification reaction for all three hydroxyl groups of uridine with fatty and aromatic acids was performed by Steglich esterification. DCC (1.5 equivalents respect to organic acid) was aggregated to organic acid (3.2 equivalents respect to uridine) solution in Chloroformo/DMF/TEA (1:2:1) mixture, after DMAP (5 mol-%) and uridine (300 mg) was added14,17,18. The reaction mixture was stirred at room temperature for 8 to 10 days (for chemical reaction see figure 1).
3',4',6'-O-tris-(o-N-Acetyl)-benzoyl-uridine (3',4',6'-O-tris-(O-N-acetyl)-anthraniloyl-uridine) (2)
The product was isolated as yellow solid (10-day reaction time, 25% yield). 1H NMR and 13C NMR data are in table 1. ESI-MS (positive mode) m/z: Calculated for C36H33N5O12: 727.6735; found, 750.5 [M+Na]+.
3',4',6'-O-trimyristoyl-uridine (3)
The product was isolated as white solid (8-day reaction time, 20% yield), Melting point: 63-64 oC. 1H NMR and 13C NMR data are in table 1. ESI-MS (positive mode) m/z: Calculated for C51H90N2O9: 875.2683; found, 898.6 [M+Na]+.
3',4',6'-tristearoyl-uridine (4)
The product was isolated as white solid (8-day reaction time, 20% yield), Melting point: 68-69 oC. 1H NMR and 13C NMR data are in table 1. ESI-MS (positive mode) m/z: Calculated for C63H114N2O9: 1043.5872; found, 1066.9 [M+Na]+ and 1044.9 [M+H]+.
3',4',6'-trioleoyl-uridine (5)
The product was isolated as yellow foam (9-day reaction time, 15% yield). 1H NMR and 13C NMR data are in table 1. ESI-MS (positive mode) m/z: Calculated for C63H108N2O9: 1037.5396; found, 1059.9 [M+Na]+.
3',4'- uridine acetonide (6)
Uridine (300 mg) was dissolved in dry acetone, after sulfuric acid was added to solution (30 uL at 98%). The reaction mixture was stirred at room temperature for 3 hours14,16. The product was isolated as yellow solid (90% yield). 1H NMR and 13C NMR data are in table 1. ESI-MS (positive mode) m/z: Calculated for C12H16N2O6: 284.2652; found, 307.1 [M+Na]+.
Viability assay
CHO-K1 cell line (Chinese Hamster Ovary, ATCC No CCL61) and MCF-7 (human breast adenocarcinoma, ATCC No HTB22), Ham F12 (Sigma) and DMEM (Sigma) culture medium, fetal bovine serum (FBS, Gibco, Brazil), penicillin 100 U/ml-streptomycin 100 µg/mL (Gibco) and (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide,thiazolyl blue) (MTT, Sigma). Solvents: DMSO reagent grade 99,9% purity (Sigma) and 2-propanol reagent grade 98% purity (J. T. Baker). Absorbance was read in Multiskan FC Microplate Photometer-ThermoScientific spectrophotometer.
Cell culture
CHO-K1 and MCF-7 cell lines in exponential phase were cultured in F-12 or DMEM medium were supplemented with 5% fetal bovine serum (FBS), penicillin 100 U/mL and streptomycin 100 µg/mL. The cultures were incubated at 37oC during 48 hours. MCF-7 cell line was incubated in wet atmosphere with 5% CO2.
MTT assay
MCF-7 and CHO-K1 cells were seeded in 96 well plates with a cell density 5x103 and 6x103 cells per well respectively, in DMEM or F-12 medium were supplemented with 5% FBS. The cultures were treated with concentrations of 1, 10 and 100 μM of uridine or derivatives, were incubated for 48 hours at 37oC for 48 hours. Later, 10 μL of MTT (5 mg/mL, Sigma) were added, and the plates were incubated in darkness at 37oC for 4 hours. After 100 μL of acidified isopropanol were added to plates, the cultures were agitated at room temperature for 1 hour. Absorbance was measured at 570 nm and cell viability percentage was calculated in three replicas of each treatment. Results were expressed with mean and standard deviation (X±SD) in at least two experiments. Statistical analysis was performed with two-way ANOVA, Bonferroni test with p<0.05 with GraphPad Prism Version 5.0 Software.
Results
Chemical
The Steglich esterification does not require of heating or an aggressive medium (acidic or basic), therefore, it is ideal method to work substrates highly acid labile or thermo-labile as uridine. This method allowed the esterification of all three hydroxyl groups of uridine, with conversions 55-60% with fatty acids and about 50% for aromatic acid, but the tri-acyl ester derivatives of uridine were obtained in lower yields 15-25%.
The tri-acyl ester derivatives were identified by comparison of 1H-NMR, 13C-NMR, 1H-1H COSY, HSQC and HMBC data of uridine and uridine acetonide. 1H-NMR and 13C-NMR signals for ribofuranose ring protons from derivatives, were deviated significantly of the substrate (uridine) and acetonide derivative (see table 1). The NMR data are agreement with the proposed structures.
Viability assay
Figure 2, shows the results of cell viability obtained by MTT assay with uridine, tri-esterified derivatives and acetonide. The uridine and its derivatives, to the lower concentrations evaluated, did not present significant inhibition on cell viability in CHO-K1 and MCF-7 cells (1 μM and 10 μM), a reduction in the cell viability was observed on both lines, when the nucleoside and derivatives were assessed to the maximum concentration (100 μM). However, only three derivatives (acetonide and compounds 2 and 3) showed significant inhibition on cellular viabilities of CHO-K1 cell line (99, 80, 30%, respectively) and MCF-7 cell line (90, 30, 30%, respectively). The uridine to the highest concentration tested (100 μM), diminished the cell viability of MCF-7 cells around 20% compared.

Discussion
Chemical
The low yields for tri-acyl ester derivatives possibly are due to three reasons: The first, the Steglich esterification requires the formation of O-acyl-isourea intermediate, starting of N,N'-dicyclohexyl-carbodiimide and the carboxylic acid, this intermediate presents a reactivity similar to the corresponding carboxylic anhydride, however, the fatty O-acyl-isourea intermediate is a bulky compound, the steric effects cause a decrease of effective shocks between the intermediate and nucleophilic catalyst (DMAP), therefore the reaction rate and yields are low14,17,18. The aromatic O-acyl-isourea intermediate is weakly activated, this showed less reactivity and higher stability, due to the π-density delocalization between aromatic and carbonyl system. Therefore, alone a uridine derivative with aromatic acids was obtained17.
Another reason is due the steric effects presented in the uridine, between the aromatic or fatty acyl ester groups (adjacent to hydroxyl) and acyl group of quaternary amide intermediate (intermediate formed by reaction among O-acyl-isourea and DMAP), when the hydroxyl is approaches to carbonyl of intermediate14,17,19. Finally, in the reactions carried out with a solvents mix of mean polarity, these solvents prevents that substrate dissolves completely in the organic phase, therefore the effective collisions between the substrate and intermediate 2 are decreased (quaternary acyl amide).
The COSY H-H spectrum for 3',4',6'-O-trimyristoil-uridine in CDCl3 presents a coupling by multiple of H-5' and H-6' protons of ribose and a multiplet with high deshielding, δ5.36 ppm appears for H-3' and H-4' protons of ribose (they protons are overlapped), at the same time, this multiple is coupled with a highly deshielded doublet, δ=6.08 ppm appears for the H-2' proton (the anomeric proton). However, the H-3' and the H-4' protons of the ribose in the spectrum for 3',4',6'-O-trimyristoyl-uridine in DMSO d-6, are not overlapped (see table 1 and figure 1). In down-field is observed a coupling by the doublet δ=5.80 ppm appears for the H-5 proton of uracil and the doublet with highest deshielding δ=7.45 ppm, appears for the H-6 proton of uracil (see table 1).
The HSQC spectrum for 3',4',6'-O-trimyristoyl-uridine in CDCl3, presents two couplings by the multiplet of the H-5' and H-6' protons of ribose with two kinds of carbons: a highly shielding carbon, δ=62.61 ppm, appears for the C-6' (-CH2-) carbon of ribose and another carbon with high deshielding δ=80.18 ppm, appear for the C-5' (-CH-) carbon of ribose. Besides, it is observed other two couplings between the multiplete of the H-3' and H-4' protons of ribose and two signals of carbons: a carbon with chemical shift δ=70.05 ppm, appears for the C-4' carbon of ribose and other carbon with higher deshielding (δ=72.61 ppm) appears for the C-3' carbon of ribose (see table 1). Furthermore, this showed a coupling by the anomeric proton (position H-2' of ribose) with a carbon high deshielding, δ=87.17 ppm appear for the C-2' carbon of ribose. In down-field, is observed a coupling by doublet for H-5 proton of uracil and a carbon with high deshielding δ=103.16 ppm, carbon of the same position. Finally, the spectrum presents a coupling between the proton with highest deshielding (position H-6 of uracil) and a highly deshielded carbon, δ=139.12 ppm, appears for the C-6 carbon of uracil (see table 1). Similarly the other compounds were analyzed.
The HMBC spectrum for 3',4',6'-O-trimyristoyl-uridine in CDCl3, presents a coupling by multiplete of H-3' and H-4' protons of ribose and a signal from highly deshielded quaternary carbons, δ=172.46 ppm appear for the ester carbonyls. Other similary coupling is observed that the multiplet for the H-6' proton of ribose and the highest deshielding quaternary carbon, δ=174.11 ppm appears for a ester carbonyl (see table 1). In down-field, the spectrum presents two coupling between the doublet for the H-5 proton of uracil and two kinds carbon, a highly shielding carbon, δ=139.12 ppm appear for C-6 carbon of uracil, and other carbon with high deshielding, δ=157.18 ppm appears for the C-4 carbon of uracil (carbonyl of amide type of a α,β-unsaturated carbonyl system). Finally, two coupling is observed by the proton with highest deshielding (H-6 proton of uracil) is coupled with two kinds carbon, a carbon with high shielding, δ=103.16 ppm appear for C-5 carbon of uracil and other carbon with higher deshielding, δ=157.18 ppm appear for the C-4 amide carbonyl type (see table 1). Similarly the other compounds were analyzed.
Viability assay
The cytotoxic activity results (No significant effect on cell viability) obtained by uridine and derivatives, possibly indicate a resistance to the nucleoside for the MCF-7 cell line. Possibly due, first, the enzymatic targets (pyr1-3 and pyr5,6) may present a overproduction, these generally participate in the novo synthesis of pyrimidine nucleosides, the enzymatic overproduction nullifies the nucleoside savage route20. Second, the transmembrane transport channels of nucleosides (phosphoglycoprotein) can to be overexpressed, this process can cause a reduction in the nucleoside incorporation since exterior of cell21. And third, the overexpression of receptors of tyrosine kinase (ErbB) can accelerate of the cell cycle and deactivate the stop of cell cycle performed by nucleosides2.
The nucleoside acetonide and derivatives 2 (3',4',6'-o-tris-(o-n-acetyl)-benzoyl-uridine) and 3 (3',4',6'-o-trimyristoyl-uridine) showed higher inhibition effect on viability of MCF-7 cell line than uridine, this could to be due to best permeability across the cell membrane, provided by substitutes and lower stability among esterase enzymes than the other derivatives, due to a little higher repulsion among adjacent chains, therefore, the hydrolysis of 6'-acyl ester groups can to be viable and thermodynamically favorable, and it is allowed posterior phosphorylation of the OH-6' hydroxyl group9,17,19. No similar behavior for the compound 1 (3',4',6'-o-triacetyl-uridine) and compound 4 (3',4',6'-tristearoyl-uridine) was observed, possibly due that the first, can to be more labile and easily degraded, and the second, maybe presents a great steric impediment and high stability among esterase enzymes9,17,19.
Conclusions
Steglich esterification method allowed obtaining uridine tri-esterified derivatives. The three hydroxyl groups esterification, was possible due that the OH-3' and OH-4' position are syn among themselves, and anti respect to the uracil ring and other hydroxyl group (position 6'), therefore, aliphatic chains move away like scissor. Steglich esterification method using aromatic acids, showed low efficiency possibly due to a great stability exhibited by the O-acyl-isourea intermediate.
No significant effect on cell viability for the uridine and derivatives was observed with p<0.05, these substances did not present a dose-response relationship. However, the compound 3',4'-uridine acetonide, showed a differential effect and a higher activity over cell viability on MCF-7 human cancer breast cell line than for the CHO-K1 cell line. Breast cancer cell line MCF-7 employed, according to results, possibly presented resistance to uridine nucleoside.
Some of the uridine derivatives, particularly with fatty acids are too bulky, this property can inhibit the hydrolysis of 6'-acyl-ester gruop of the ribose and produce decreassed of phosphorylation (activation) of the nucleoside.
Acknowledgments
Authors thank the professor Felipe Otalvaro of the Institute of Chemistry of the University of Antioquia and young researchers: Diana M. Mejía C., Suly S. Villa V., Angélica M. Bustamante and Álvaro J. García O
Competing interest
Authors state that does not exist conflict of interests in this work.
References
1. Chik F, Machnes Z, Szyf M. Synergistic anti-breast cancer effect of a combined treatment with the methyl donor s-adenosyl methionine and the DNA methylation inhibitor 5-aza-2´-deoxycytidine. Carcinogenesis. 2014; 35(1): 138-44. [ Links ]
2. Strasser S, Maier S, Leisser C, Saiko P, Madlener S, Bader Y, et al. 5-fdurd-AraC heterodinucleoside re-establishes sensitivity in 5-fdurd- and AraC-resistant MCF-7 breast cancer cells overexpressing erbb2. Differentiation 2006; 74(9-10): 488-98. [ Links ]
3. Silvestris N, Cinieri S, La Torre I, Pezzella G, Numico G, Orlando L, et al. Role of gemcitabine in metastatic breast cancer patients: A short review. Breast 2008; 17(3): 220-6. [ Links ]
4. Mehta DR, Foon K a, Redner RL, Raptis A, Agha M, Hou J-Z, et al. Fludarabine and cytarabine in patients with acute myeloid leukemia refractory to two different courses of front-line chemotherapy. Leuk Res. 2011; 35(7): 885-8. [ Links ]
5. Bzowska A, Kulikowska E, Shugar D. Purine nucleoside phophorylases: properties, functions and clinical aspects. Pharmacol Ther. 2000; 88(3): 349-425. [ Links ]
6. Zhenchuk A, Lotfi K, Juliusson G, Albertioni F. Mechanisms of anti-cancer action and pharmacology of clofarabine. Biochem Pharmacol. 2009; 78(11): 1351-9. [ Links ]
7. Wagner CR, Iyer V V, Mcintee EJ. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med Rev Res. 2000; 20(6): 417-51. [ Links ]
8. Sarpietro MG, Ottimo S, Giuffrida MC, Rocco F, Ceruti M, Castelli F. Synthesis of n-squalenoyl cytarabine and evaluation of its affinity with phospholipid bilayers and monolayers. Int J Pharm. 2011; 406(1-2): 69-77. [ Links ]
9. Moysan E, Bastiat G, Benoit J. Gemcitabine versus modified gemcitabine: a review of several promising chemical modification. Molecular pharmaceutics. 2013; 10: 430-44. [ Links ]
10. Yamamoto T, Koyama H, Kurajoh M, Shoji T, Tsutsumi Z, Moriwaki Y. Biochemistry of uridine in plasma. Clin Chim Acta. 2011; 412(19-20): 1712-24. [ Links ]
11. Wandzik I, Bieg T, Czaplicka M. Synthesis of 2-deoxy-hexopyranosyl derivatives of uridine as donor substrate analogues for glycosyltransferases. Bioorg Chem. 2009; 37(6): 211-6. [ Links ]
12. Valade A, Urban D, Beau J-M. Target-assisted selection of galactosyltransferase binders from dynamic combinatorial libraries. An unexpected solution with restricted amounts of the enzyme. Chembiochem. 2006;7(7): 1023-7. [ Links ]
13. Hrdlicka P.J, Jepsen J.S, Nielsen C, Wengela J. Synthesis and biological evaluation of nucleobase-modified analogs of the anticancer compounds 3'-C-ethynyluridine (Eurd) And 3'-C-ethynylcytidine (Ecyd). Bioorg. Med. Chem. 2005; 13: 1249-60. [ Links ]
14. Berrío Escobar JF, Pastrana Restrepo MH, Mejía Cuartas DM, Márquez Fernández DM, Márquez Fernández ME, Martínez Martínez A. Síntesis y actividad citotóxica de conjugados de la uridina con triterpenos en celulas de cancer de mama. Ars Pharm. 2016; 57(2): 55-62. [ Links ]
15. McEvillyn M, Popelas C, Tremmel B. Use of uridine triacetate for the management of fluorouracil overdose. Am J Health-Syst Pharm. 2011; 68:1806-09. [ Links ]
16. Clayden J, Greeves N, Warren S. and Wothers P. Organic Chemistry. 2dn Ed. Oxford: Oxford University Press. 2012. 1234 p. [ Links ]
17. Radi M, Adema AD, Daft JR, Cho JH, Hoebe EK, Alexander LMM, et al. In Vitro Optimization of Non-Small Cell Lung Cancer Activity with Troxacitabine. J. Med. Chem. 2007; 50(7): 2249-53. [ Links ]
18. Neises B, Steglich W. Simple method for the esterification for carboxylic acids. Angew Chem Int. 1978; 17(7): 522-4. [ Links ]
19. Chhikara BS, Mandal D, Parang K. Synthesis and evaluation of fatty acyl ester derivatives of cytarabine as anti-leukemia agents. Eur J Med Chem. 2010; 45(10): 4601-8. [ Links ]
20. Karle JM, Cowan KH, Chisena CA, Cysyk RL. Uracil Nucleotide Synthesis in a Human Breast Cancer Cell Line (MCF-7) and in Two Drug-Resistant Sublines that Contain Increased Levels of Enzymes of the de Novo Pyrimidine Pathway. Molecular Pharmacology. 1986; 30: 136-41. [ Links ]
21. Morgan PE, Fine RL, Montgomery P, Marangos PJ. Multidrug resistance in MCF-7 human breast cancer cells is associated with increased expression of nucleoside transporters and altered uptake of adenosine. Cancer Chemother Pharmacol. 1991; 29: 127-32. [ Links ]
![]() Correspondence:
Correspondence:
Jhon Fernando Berrío Escobar,
Medellín-Antioquia, Colombia.
+57-4-2198456
jhon.berrio@udea.edu.co.
Received: 07.10.2016
Accepted: 18.12.2016

