










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Se ha empleado el monitoreo de medicamentos posteriores a la comercialización para juzgar la calidad, eficacia terapéutica y seguridad de la medicina. La información obtenida de dicho monitoreo podría ser utilizada para el desarrollo de productos y la mejora de la normativa vigente. En esta investigación se estudió la comparación de la calidad biofarmacéutica de tabletas de Clonazepam 0,5 mg innovador y multifuente comercializados en el mercado peruano.
El nombre químico del Clonazepam (CZP) es 1,3-dihidro-2H-1,4-benzodiazepin-2-ona en el que los hidrógenos en las posiciones 5 y 7 están sustituidos por grupos 2-clorofenilo y nitro, respectivamente. Se utiliza en el tratamiento de todos los tipos de epilepsia y convulsiones, así como en el mioclono y los movimientos anormales asociados y los trastornos de pánico. Sin embargo, su uso puede verse limitado por el desarrollo de la tolerancia y la sedación. Tiene un papel como anticonvulsivo, modulador del ácido gamma-aminobutírico (GABA) y fármaco ansiolítico. Es una 1,4-benzodiacepina y un miembro de monoclorobencenos (Figura 1). Es un compuesto básico, con un pKa de 4,9 y se encuentra casi totalmente bajo forma no ionizada (90 %) a pH fisiológico de 7,4, pudiendo de esta manera atravesar fácilmente las membranas celulares. Con respecto a sus características organolépticas, es un polvo cristalino blanquecino a amarillo, prácticamente inodoro e insípido. Por ser un derivado de las benzodiacepinas posee propiedades ansiolíticas, sedantes, hipnóticas y anticonvulsivas. Además, potencia las actividades inhibidoras del GABA al unirse al receptor GABA, ubicado en el sistema límbico y el hipotálamo. Esto aumenta la frecuencia de apertura del canal de cloruro, lo que permite el flujo de iones de cloruro en la neurona y, en última instancia, conduce a la hiperpolarización de la membrana y a una disminución de la excitabilidad neuronal. Fue sintetizado al final de los años 1960 por La Roche y se comercializa con el nombre comercial Rivotril®1.
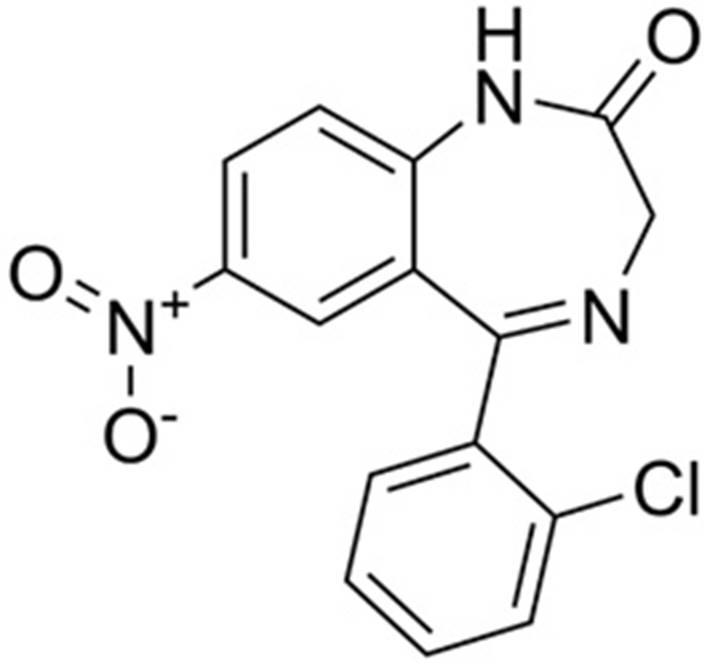
De acuerdo con el Sistema de Clasificación Biofarmacéutica (BCS) propuesto por Amidon, G. et al.(1995), las sustancias farmacológicas son clasificados en cuatro clases según su solubilidad y permeabilidad. En diferentes investigaciones realizadas por el Food & Drug Administration (FDA), concluyen que el CZP está en el BCS Clase I, estas sustancias presentan buena solubilidad en agua y además se absorben con facilidad a través de la mucosa gastrointestinal2,3,4. Por esto, su biodisponibilidad por vía oral es prácticamente del 100%, siempre y cuando no presente metabolismo presistémico ni se degrade a nivel gastrointestinal5. Para Iriarte, R.(2015), algunos medicamentos multifuente pueden estar exentos de presentar estudios de biodisponibilidad “in vivo” ya que, por sus características, pueden demostrar bioequivalencia a través de otros métodos. Estos medicamentos se comercializan por un procedimiento diferente, y se denominan bioexenciones6. Según Alfredo, L.; Campos, S.(2017), una bioexención es la autorización de comercialización de una formulación oral basándose, única y estrictamente, en criterios de disolución, en sustitución de un estudio de bioequivalencia “in vivo”7.
Usando el trabajo de Löbenberg y Amidon(2000), la FDA presentó en el año 2003 las primeras guías para la exención de estudios de bioequivalencia “in vivo”, para las formas farmacéuticas orales de liberación inmediata pertenecientes a la clase I del sistema BCS (alta solubilidad y alta permeabilidad)8. Esta iniciativa permitía demostrar la bioequivalencia mediante unos ensayos de disolución “in vitro”9.
Al principio, las bioexenciones eran usadas por los propios fabricantes de los medicamentos de referencia cuando necesitaban realizar cambios post-registro, o cambios de escala que no afectaban a la indicación de la formulación10.
Para Shah et al. 2016, el riesgo de una discrepancia terapéutica por errores de bioequivalencia siempre existe al usar un medicamento genérico, incluso si se lleva a cabo un ensayo clínico completo. La autorización de un medicamento por bioexención se realiza basándose en estadísticas y datos científicos representativos del producto11. Según Baishya et al. 2018, actualmente existe un comportamiento psicológico común de que los productos farmacéuticos de alto costo fabricados por las principales compañías farmacéuticas son mejores en comparación con los productos de bajo costo fabricados por pequeñas empresas a escala12. Hasta la fecha no existen suficientes estudios de este medicamento usado frecuentemente para los casos clínicos anteriormente mencionados. Estos hechos dirigieron el interés para evaluar la calidad de algunas marcas de CZP disponibles comercialmente en el mercado peruano con especial énfasis en la desintegración y estudio de disolución debido a su enorme prominencia prediciendo la biodisponibilidad y la calidad del producto. Seis unidades de cada marca se utilizaron para la disolución. Otros parámetros de calidad para las tabletas se realizaron como dureza, friabilidad, variación de peso, tiempo de desintegración, cuantificación de principio activo y uniformidad de contenido también fueron determinados de acuerdo con los protocolos establecidos. Los resultados de la prueba fueron sometidos a análisis estadísticos para comparar el Perfil de disolución. Modelos de enfoques independientes de factor de diferencia (f1), factor de similitud (f2) y Eficiencia de disolución (% ED) también fueron empleados13.
Métodos
Materiales
Todos los productos químicos utilizados fueron comprados de Merck Company. Las tabletas de Clonazepam 0,5 mg (de fabricación nacional e importado) se compraron en las diferentes boticas autorizadas del mercado local. El estándar de Clonazepam fue adquirido de E. Merck, Darmstadt, Alemania. Las pruebas de cromatografía de líquidos de alto rendimiento se realizaron en un HPLC-20 AT pump, SPD-20 A UV/visible detector (Shimadzu, Japón). Empleando membranas Millipore(0,45) fabricadas en USA. Utilizando equipos para medir la friabilidad, la dureza, el tiempo de desintegración y la disolución como: el medidor de friabilidad (Veego friabilator VFT-2, USA), los aparatos de disolución (TDT-08L, Electrolab, USA), el probador de dureza (8M, Dr. Schleuniger, Switzerland), el aparato de desintegración (Model: VDT-2, Veego USA) y balanza analítica (Model: AY-200, SHIMADZU Corporation, Japan). Se utilizó para medir la variación de los pesos de las tabletas.
Medición de la dureza de las tabletas de Clonazepam (prueba de dureza)
Sobre la base del método publicado en la USP 42, se tomaron por separado 20 tabletas de cada uno de los tipos utilizados en esta investigación Clonazepam innovador (CZPR), Clonazepam multifuente 1 (CZPM1) y Clonazepam multifuente 2 (CZPM2). El grado de dureza de cada tipo se midió mediante el siguiente procedimiento: Poner en funcionamiento el equipo, se seleccionó la unidad de trabajo para la dureza “(kp)” de kilopondios. Al mismo tiempo, la barra medidora se abrió para dar campo a la tableta a ensayar, retirando el protector plástico y colocando la primera tableta entre la barra estacionaria y la barra medidora (movible). La barra medidora movible inició su movimiento horizontal hacia la barra estacionaria de modo que presionó la tableta hasta provocar su ruptura. En ese momento en la pantalla se visualizó el valor de la dureza obtenida de cada tableta. La barra medidora se abre automáticamente, dando el espacio necesario para proceder a limpiar la zona, retirar el protector plástico y con la ayuda de una brocha eliminar la tableta rota. Una vez realizada la limpieza, se colocó la siguiente tableta a ensayar procediendo como se indica desde el segundo punto14.
Medición de la friabilidad de las tabletas de Clonazepam (prueba de friabilidad)
Sobre la base de los métodos informados en la USP 42, para tabletas con un peso unitario igual o menor a 650 mg, se tomó una muestra de tabletas enteras correspondiente lo más cercano posible a 6,5 g. Para tabletas con un peso unitario mayor a 650 mg, tomar una muestra de 10 tabletas enteras. Debió quitarse el polvo de las tabletas cuidadosamente antes de realizar la prueba. Pesar con exactitud la muestra de tabletas y colocarla en el tambor. Hacer girar el tambor a 25 rpm x 4 minutos y retirar las tabletas. Quitar el polvo suelto de las tabletas como se hizo anteriormente y pesar con exactitud. Cada conjunto de tabletas se colocó simultáneamente en el instrumento Friability Tester. Se calcularon los porcentajes de friabilidad de los comprimidos. Generalmente la prueba se realiza una vez. Si se encuentran tabletas claramente agrietadas, segmentadas o rotas en la muestra de tabletas después de la prueba, se puede decir que no ha pasado la prueba. Para este caso se considera aceptable una pérdida media máxima de peso de la muestra utilizada de no más de 1,0%14.
Medición del tiempo de desintegración de las tabletas de Clonazepam
El instrumento estuvo equipado con una canasta que contuvo 6 tubos de extremo abierto con una longitud de 7,5-8 cm y un diámetro de 2,15 cm. Un tamiz de acero inoxidable de malla 10 se puso debajo de los tubos. En un vaso beaker de 1 000 mL de capacidad se colocó el medio de desintegración. El volumen del líquido en el recipiente es tal que, en el punto más alto del recorrido ascendente, la malla de alambre permanece al menos 15 mm por debajo de la superficie del líquido y desciende a no menos de 25 mm del fondo del recipiente, en el recorrido descendente. En ningún momento puede quedar sumergida la parte superior del montaje canastilla-gradilla. Verificar que el medio especificado como líquido de inmersión (agua purificada) este a 37 °C ± 2 ºC. Controlar la temperatura del baño de agua manteniendo el termómetro sumergido durante todo el ensayo. Cada vez, se tomaron seis tabletas al azar de cada grupo de muestra y a cada tubo de vidrio de extremo abierto, se colocó una tableta y se cubrió con una lámina de plástico de 9,35-9,65 mm de espesor y 20,55-20,85 mm de diámetro. La lámina está hecha de un material plástico transparente adecuado, con un peso específico entre 1,18-1,20. Luego, se encendió el instrumento y se registró el tiempo de desintegración de cada tableta. Se determinaron y registraron el tiempo de desintegración mínimo, máximo y promedio de cada tipo de tableta. El tiempo mínimo de desintegración corresponde al momento en que la primera tableta comenzó a desintegrarse y el tiempo máximo de desintegración fue el momento en que la última tableta comenzó a desintegrarse. De acuerdo con la regla general, las seis tabletas recubiertas en agua destilada deben desintegrarse en un período de hasta 15 minutos. Se considera que las muestras se han desintegrado y se registra el tiempo de desintegración cuando: no quedan residuos en la canastilla, los residuos de la muestra (forma farmacéutica sólida evaluada) constituyen una masa blanda sin presencia de contenido (polvo, gránulos, etc.) y cuando quedan fragmentos insolubles del recubrimiento, que permanecen en la canastilla o se adhieren a la superficie del disco14.
Variación de peso
Se seleccionaron al azar 20 tabletas de cada uno de los tipos de tabletas y se pesaron con una balanza analítica precisa. De acuerdo con las referencias auténticas, el rango aceptable debe estar dentro del 92,5-107,5% del peso del medio14.
Preparación de la solución madre
Pesar con exactitud alrededor de 20 mg de ER Clonazepam (Estándar de Referencia), transferir a un matraz volumétrico de 200 mL, adicionar 50 mL de diluyente, llevar al ultrasonido hasta disolver y llevar a volumen con diluyente. Transferir 10 mL de la solución anterior a un matraz volumétrico de 50 mL, diluir y llevar a volumen con diluyente. Homogeneizar. Filtrar por membrana HVLP de 0,45 µm e inyectar (Concentración aproximada: 0,02 mg/mL de Clonazepam)14.
Preparación de las soluciones estándar
Para trazar la curva de calibración, se necesitaron concentraciones de 0,2; 0,4; 0,6; 0,8 y 1,0 mg/mL. De la solución madre mencionada anteriormente, se tomaron 2, 4, 6, 8 y 10 mL y cada uno se colocó en un matraz volumétrico individual de 10 mL, luego se llevaron a volúmenes exactos de 10 mL agregando la solución de agua purificada: metanol: tetrahidrofurano (60:52:13 V/V) a cada uno de los matraces. Por lo tanto, se obtuvieron soluciones con concentraciones de 0,2; 0,4; 0,6; 0,8 y 1,0 mg/mL que se usaron para trazar la curva de calibración e inyección en el instrumento de HPLC14.
Elaboración de la curva de calibración del estándar
Para elaborar la curva estándar, se inyectaron 5 veces y cada vez 20 µL de cada una de las soluciones estándar preparadas en el equipo de HPLC desde las concentraciones más bajas a las más altas. Los cromatogramas y los datos relevantes, como el área del pico, la altura del pico, el tiempo de retención, etc., se registraron y guardaron como tablas de informe de pico en el programa del software. Para garantizar la precisión del método de medición de todos los procedimientos para elaborar la curva de calibración se repitieron tres veces. Luego, se trazó la curva de calibración. Sobre la base de la curva de calibración, las muestras desconocidas se inyectaron en el instrumento de HPLC y los cromatogramas se registraron, luego se determinaron las cantidades de activo de las muestras desconocidas14.
Cuantificación del principio activo
Tomar no menos de 20 tabletas y moler a polvo fino. Pesar con exactitud alrededor de 3200 mg de muestra (equivalente a 10 mg de Clonazepam), transferir a un matraz volumétrico de 100 mL, agregar 75 mL de diluyente y llevar a ultrasonido la muestra por 30 minutos con agitación intermitente cada 5 minutos y evitando que se caliente, atemperar y llevar a volumen con diluyente. Homogeneizar y filtrar por membrana HVLP de 0,45 µm e inyectar (Concentración aproximada: 0,1 mg/mL de Clonazepam). Las Tabletas de Clonazepam 0,5 mg deben contener no menos de 90,0% y no más de 110,0% (± 0,05 mg) de la cantidad declarada14.
Uniformidad del contenido
Se utilizó 10 comprimidos de cada tipo. Para este ensayo se agregó una tableta (equivalente a 0,5 mg de Clonazepam) a un matraz volumétrico de 25 mL, agregar 10 mL de diluyente y agitar manualmente procurando la humectación de la tableta y llevar al ultrasonido por 20 minutos con agitación intermitente cada 5 minutos, atemperar y llevar a volumen con diluyente. Homogeneizar y filtrar por membrana de 0,45 µm e inyectar (Concentración aproximada: 0,02 mg/mL de Clonazepam)14.
Medición de la velocidad y perfil de disolución
De acuerdo con las condiciones informadas en la monografía de Clonazepam en la Farmacopea de la USP 42, se usó 900 mL de agua desgasificada como medio de disolución. Las mediciones se realizaron mediante un instrumento de disolución tipo cesta con 75 rpm. La temperatura del medio se ajustó a 37°C ± 0,5 °C. Según la farmacopea de USP 42, el 75% del ingrediente activo de la tableta debe disolverse en el medio después de 45 minutos. En esta investigación, se colocaron individualmente 6 comprimidos de cada tipo en la canasta especial del instrumento y se midió y calculó la extensión de la velocidad de disolución como porcentaje del ingrediente activo liberado en varios momentos. Después del inicio de la prueba, se extrajo una alícuota de 20 mL del medio de disolución en intervalos de tiempo de 5, 10, 15, 20, 30, 45 y 60 minutos, y se filtró individualmente por membrana de PVDF (hidrófila) de 0,45 μm y de 25 mm. Mientras tanto, después de cada extracción de la alícuota de 20 mL, se añadieron 20 mL de agua desgasificada al medio de disolución para obtener el volumen hasta 900 mL. La cuantificación del fármaco fue realizada a través de método cromatográfico de HPLC. Las condiciones utilizadas para el análisis fueron: fase móvil agua:metanol:acetonitrilo 40:30:30, 20 μL de volumen de inyección, flujo de 1 mL/min, columna C18 4 mm x 30 cm L1, y detector a 254 nm.14. Se determinó el grado de disolución como el porcentaje liberado del ingrediente activo. Al dividir los mg del posible ingrediente activo de una tableta de Clonazepam (0,5 mg) en el volumen total del medio de disolución (900 mL), se obtuvo una concentración de 11,1 µg/mL. Esta concentración fue considerada como 100% de liberación de fármaco. Al dividir las concentraciones obtenidas a partir de los datos de HPLC para cada tableta individual en los tiempos especificados, se calculó el porcentaje de liberación para cada tableta14.
Eficiencia de disolución (ED%)
La eficiencia de disolución (ED) se empleó para comparar la liberación del fármaco de las diferentes marcas. La eficiencia de disolución es el área bajo la curva de disolución dentro de un rango de tiempo expresado como un porcentaje de la curva de disolución al máximo durante el mismo período de tiempo. Se puede decir que el medicamento innovador y el producto multifuente son equivalentes si la diferencia entre sus eficiencias de disolución está dentro de los límites apropiados (± 10%, que a menudo se usa)15.
Resultados
Tabla 1. Dureza promedio (KP) y Velocidad de desintegración (min) de los comprimidos innovador y multifuente de Clonazepam 0,5 mg.
| CLONAZEPAM | PROMEDIO (KP) | D.S. | PROMEDIO (MIN) | D.S. |
|---|---|---|---|---|
| CZPR | 4,4 | 0,19 | 5,3 | 0,10 |
| CZPM1 | 4,5 | 0,28 | 5,5 | 0,05 |
| CZPM2 | 4,5 | 0,28 | 5,8 | 0,12 |
Tabla 2. Peso promedio de los comprimidos innovador y multifuente de Clonazepam 0,5 mg.
| CLONAZEPAM | PROMEDIO (mg) | C.V. | D.S. | VARIANZA |
|---|---|---|---|---|
| CZPR | 89,8 | 1,73% | 1,55 | 2,42 |
| CZPM1 | 89,9 | 1,63% | 1,46 | 2,14 |
| CZPM2 | 90,3 | 0,87% | 0,78 | 0,61 |
Tabla 3. Especificación establecida del porcentaje peso perdido después del ensayo de friabilidad según USP 42 para los comprimidos innovador y multifuente de Clonazepam 0,5 mg.
| CLONAZEPAM | PESO INICIAL (mg) | PESO FINAL (mg) | % DE PERDIDA | ESPECIFICACIÓN USP 42 | CUMPLE |
|---|---|---|---|---|---|
| CZPR | 6652,0 | 6638,7 | 0,20 | % DE PERDIDA < 1% | SI |
| CZPM1 | 6749,1 | 6714,3 | 0,52 | SI | |
| CZPM2 | 6697,3 | 6670,2 | 0,40 | SI |
Tabla 4. Porcentaje promedio del contenido de principio activo según lo declarado (%) y Porcentaje promedio de la cantidad disuelta de 6 tabletas para establecer la velocidad de disolución (Q%) de los comprimidos innovador y multifuente de Clonazepam 0,5 mg.
| CLONAZEPAM | PROMEDIO (%) | D.S. | Q% (PROMEDIO) | D.S. |
|---|---|---|---|---|
| CZPR | 101,81 | 0,39 | 99 | 1,21 |
| CZPM1 | 100,13 | 0,39 | 100 | 1,05 |
| CZPM2 | 99,87 | 0,28 | 99 | 1,03 |
Tabla 5. Factor de Diferencia (f1) y Factor de similitud (f2) de CZPR con los CZPM1 y CZPM2.
| f1 | CZPM1 | CZPM2 |
|---|---|---|
| CZPR | 7,86 | 4,45 |
| f2 | CZPM1 | CZPM2 |
| CZPR | 58,40 | 69,96 |
Tabla 6. Perfil de disolución y Eficiencia de disolución (ED%) del CZPR, CZPM1 y CZPM2.
| TIEMPO | CZPR | CZPM1 | CZPM2 | |||
|---|---|---|---|---|---|---|
| MEDIA | D.S. | MEDIA | D.S. | MEDIA | D.S. | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | 77,01 | 10,02 | 83,00 | 5,49 | 92,48 | 6,82 |
| 10 | 85,78 | 10,08 | 93,57 | 5,01 | 105,97 | 1,42 |
| 15 | 90,13 | 10,37 | 98,75 | 4,41 | 108,44 | 1,99 |
| 20 | 92,32 | 11,68 | 100,36 | 4,00 | 108,68 | 1,83 |
| 30 | 94,60 | 11,19 | 101,23 | 3,38 | 108,07 | 1,82 |
| 45 | 96,67 | 11,32 | 103,43 | 4,51 | 108,35 | 1,67 |
| 60 | 97,43 | 10,92 | 103,25 | 3,60 | 107,21 | 1,57 |
| ED% | 88,67 | 95,36 | 92,66 | |||

Discusión
Los estudios de bioequivalencia in vitro están constituidos por estudios comparativos de perfiles de disolución y pruebas fisicoquímicas, en donde se determina la cantidad o porcentaje del principio activo disuelto en función del tiempo bajo condiciones controladas y validadas. Para productos farmacéuticos altamente solubles, y altamente permeables, la bioequivalencia in vitro (estudios de disolución) es apropiada y considerada como criterio necesario y suficiente para comparar los medicamentos innovador y multifuentes. El uso de estas técnicas ha permitido no exigir bioequivalencia in vivo para un número importante de medicamentos16.
Todas las tabletas de Clonazepam 0,5 mg utilizadas en esta investigación estaban dentro de su vida útil de 3 años a partir de la fecha de fabricación. Los comprimidos fueron sometidos a una serie de pruebas para evaluar los parámetros de calidad. No se encontraron anomalías en la apariencia física de las muestras de diferentes marcas.
Las muestras obtenidas del mercado local fueron sometidas a una serie de pruebas para evaluar los parámetros de calidad. La prueba de dureza del material es indicativa de su resistencia. La característica física más importante para evaluar la tableta es la dureza17. El límite aceptable de dureza de una tableta es de 5 a 8 Kp. Además, una fuerza entre 4 y 10 Kp también se considera satisfactoria18. Se determinó la dureza de las diferentes tabletas de Clonazepam y estaba entre 4,4 y 4,5 Kp que cumple con la especificación USP (Tabla 1).
El tiempo de desintegración de las tabletas de Clonazepam 0,5 mg se muestra en la Tabla 1. Ninguna de las muestras excedió la especificación del tiempo de desintegración que estaba entre 5,3 y 5,8 min.
La prueba de variación de peso es un método satisfactorio para determinar la uniformidad del contenido del medicamento de las tabletas y sirve como un indicador de las buenas prácticas de manufactura (BPM) mantenidas por los fabricantes, así como la cantidad de ingrediente farmacéutico activo (IFA) contenido en la formulación19. La variación de peso para todas las tabletas utilizadas en este estudio mostró cumplimiento dentro de las especificaciones oficiales, ya que ninguno de los productos se desvió de la especificación. Cuando la variación de peso está dentro de las especificaciones, se cree que las tabletas contienen un ingrediente activo uniforme para dar la respuesta terapéutica deseada, pero cuando la variación de peso está fuera de la especificación, se piensa que las tabletas contienen menos o más ingrediente activo para dar una respuesta terapéutica ineficaz o efecto tóxico respectivamente. Puede variar debido al resultado de las deficientes propiedades de flujo de granulación, lo que resulta en un relleno desigual de la matriz18. Se observó que todas las marcas cumplen con la especificación USP que estaba entre 89,8 mg ± 1,55%, 89,9 mg ± 1,46% y 90,3 mg ± 0,78% (Tabla 2).
Los resultados de la prueba de friabilidad son inferiores al 1%, y según la farmacopea, el límite es del 1% del peso inicial de las tabletas, por lo que todas las tabletas de Clonazepam 0,5 mg pasaron a la prueba de friabilidad, lo que significa que todas estas marcas de tabletas de Clonazepam tienen buena resistencia y puede tolerar los golpes durante el transporte de las mismas (Tabla 3).
Los resultados de las pruebas de dosaje de todas las marcas fueron de entre 99,87 y 101,81% que cumplen con la especificación USP 42 para la prueba de ensayo (Tabla 4).
Se determinó la velocidad de disolución de las diferentes tabletas de Clonazepam 0,5 mg. La especificación USP es mayor a 85% de la cantidad declarada, los resultado mostrados en la tabla 4 indican que la disolución entre el 99 y 100% se encuentra dentro de lo establecido por la farmacopea.
Para comparar los perfiles de disolución de las tabletas, se empleó un enfoque independiente del modelo de factor de diferencia (f1) y factor de similitud (f2).
El factor de diferencia (f1) es el porcentaje de diferencia entre dos curvas en cada punto y es una medida del error relativo entre las dos curvas. El factor de similitud (f2) es una transformación de raíz cuadrada recíproca logarítmica de la suma del error al cuadrado y es una medida de la similitud en el porcentaje (%) de disolución entre dos curvas19. La Tabla 5 muestra los valores f1 y f2 de diferentes marcas con respecto a la marca innovadora elegida.
En el cálculo de f2, generalmente solo se considera una medición después de que el producto comparador haya alcanzado el 85% de disolución.
Los valores para f2 son más de 50 y todos los valores de f1 son menos de 15. Por lo tanto, podemos decir que todas las marcas son equivalentes con el producto innovador (Tabla 5).
La eficiencia de disolución (ED) también se empleó para comparar la liberación del fármaco del medicamento innovador y multifuentes. La eficiencia de disolución es el área bajo la curva de disolución dentro de un rango de tiempo. Algunos estudios de bioequivalencia indican que cuando la diferencia de eficiencia de disolución del medicamento innovador con el multifuente no difiere de ± 10% se consideran como equivalentes20,21. La ED de los multifuentes no difirió en 10% con la marca innovadora. Entonces, podemos decir que todos los medicamentos multifuentes en estudio son equivalentes a la marca innovadora.
Los resultados del estudio del perfil de disolución muestran que las diferentes marcas liberan aproximadamente el 100% del medicamento en una hora. Todas liberan más del 70% del principio en 5 minutos (Tabla 6), lo que significa que el principio activo, se disuelve rápidamente en estas marcas. Los datos demuestran que el estudio de disolución de Clonazepam cumple con los estándares de la farmacopea y al no existir diferencia significativa en los diferentes tiempos de muestreo, nos permiten establecer que pueden intercambiarse los medicamentos multifuente de Clonazepam con el innovador (Figura 2).
Conclusiones
En la práctica industrial actual, para comparar el medicamento innovador con los multifuentes, las pruebas in vitro juegan un papel importante. Esta investigación revela que no hay diferencias notables en los parámetros de calidad de los productos investigados. Los datos presentados demuestran que los multifuentes de Clonazepam 0,5 mg comparados con el innovador cumplen con los parámetros de bioequivalencia e intercambiabilidad.

