










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
Cephalexin monohydrate is a derivate of amynodesacetoxycephalosporane acid, belongs to β-lactamic antibiotics of the I generation with a range of pharmacological activity. Cephalexin has a great Gram-negative and Gram-positive, antimicrobial properties. It is produced in gelatin capsules and in the form of suspension. Cephalexin was included into the “Top 200 Drugs” mostly prescribed in the USA according to National Data Corporation Health1.
The British Pharmacopeia and the State Pharmacopeia of Ukraine recommend a potentiometric nonaqueous method for the determination of Cephalexin-standard using perchloric acid as the titrant and HPLC method using a mixture of phosphate buffer, methanol and acetonitrile as mobile phase with UV detection at 254 nm2,3.
Several procedures have been reported in the literature for the analysis of Cephalexin. The reported methods include UV-spectroscopic methods and visible spectroscopic methods4,5, spectrofluorimetry6,7, chemiluminescence8, atomic absorption spectrometric methods9, polarography10-12. The impressive increase in the use of HPLC in the past thirty years did not pass β-lactam antibiotics. HPLC has been used often in all fields of β-lactam research, not only as an assay method but also as a tool for purification of the antibiotics. Due to the insolubility of these compounds in organic solvents, normal phase LC was sparingly used13,14. But, HPLC methods are quiet expensive and product a big amount of wastes including toxic organic solvents15. Some of these methods involve several manipulation steps, which are not simple for routine analysis of pharmaceutical formulations and need advanced instruments.
The direct spectrophotometric methods suffer a lack of specificity because all compounds containing the β-lactam ring absorb in the range 250-270 nm.
The official rules in pharmaceutical preparations use iodometric titration are unselective as all antibiotics, the first substances for their production as well as their degradation products are being oxidized by iodine. It is not suitable for control of the purity of the antibiotics of this group16. The peculiarity of this method is the dependence of the titer on the conditions.
Therefore, the development of precise, correct and low cost technique for drugs analysis is of a great interest. Titrimetric, kinetic-spectrophotometric method and voltammetry may serve as useful alternative to many of the previously mentioned new sensitivity, remarkable accuracy and precision and wide applicability.
The present work aim is to develop simple, rapid and cost-effective methods for the determination of Cephalexin in pure form and dosage forms using titrimetric, kinetic-spectrophotometric and voltammetric techniques. The methods used KHSO5 as a reagent. Moreover, the developed methods meet the requirements of the concept of Green Chemistry.
MATERIALS AND METHODS
All materials were of the analytical reagent grade, and the solutions were prepared with twice-distilled water. Cephalexin pure substance “Purilex” ((6R, 7R)-7-[(R)-2-amino-2-phenylacetamido]-3-metyl-8-oxo-5-tia-1-azabicyclo[4.2.0]oct-2-en-2-carbonic acid monohydrate) produced by DMS Anti-infectives Chemferm S.A., Spain: ser. B425055; assay 100.9 % (BPh); with 6.2 % content of water). Granules for suspension preparation for internal application “Cephalexin” 250 mg/5 mL, “Hemopharm” AD, Serbia, series 3130408, content: 5 mL of suspension contain 250 mg of cephalexin monohydrate, excipients. The acceptable range is 225-300 mg/5 mL.
The oxidant was KHSO5, potassium caroate in the form of a triple potassium salt of Caro’s acid, 2KHSO5 · KHSO4 · K2SO4 (Acros Organics). The choice of the reagent was determined by its rather high oxidative capacity, E0 = 1.84 V, easy availability, and satisfactory solubility in water, and also by sufficiently high stability in the use and storage.
Preparation of standard solutions
Preparation of 0.02 M potassium caroate solution. 0.615 g of 2KHSO5 ⋅ KHSO4•K2SO4, was dissolved in twice-distilled water (100 mL). The concentration of potassium caroate was controlled by iodometric titration.
Preparation of 0.01 M sodium hydroxide solution. 0.4 g of sodium hydroxide was weighted and dissolved in 100 mL of distilled water. A 1/10 dilution was made to obtain required concentration.
Preparation of 0.02 M sodium thiosulfate solution. A 0.1 M solution of sodium tiosulfate was prepared from the standard titre fixanal. A 2/10 dilution was made to obtain required concentration.
Preparation of 5 % potassium iodine solution. 5.0 g of sodium hydroxide was weighted and dissolved in 100 mL of distilled water.
Preparation of 0.1 M sulfuric acid solution. The solution was prepared from the standard titre fixanal in a 500 mL volumetric flask.
Polarograms were recorded in a 0.03 M supporting acetate buffer solution with pH 4.5. To prepare it, 23.0 g of CH3COONa ⋅ 3H2O of chemically pure grade were dissolved in 50 mL of twice-distilled water, and pH 4.5 was set using 0.1 M HCl; the mixture was diluted to 500 mL with twice-distilled water and stirred carefully
Preparation of cephalexin working standard solutions
Seven solutions of the following concentrations: 80%, 85%, 90%, 95%, 100%, 110%, 120% were prepared. A precisely weighed portion (0.2388 g; 0.2538 g; 0.2687 g; 0.2836 g; 0.2985 g; 0.3284 g; 0.3582 g correspondingly) of the substance was transferred into a 100-mL volumetric flask and diluted to the mark.
Apparatus
Spectrophotometry
A spectrophotometer SF-46 (LOMO, JSC, St-Petersburg, Russia). 1 cm quartz cells were used for spectral measurements.
Voltammetry
Voltammograms were recorded on a CLA-3 oscillographic polarography (Rostov-on-Don Research Institute, Russia) in a three-electrode thermostated cell at 20°C; the indicator electrode was dropping mercury one, the reference electrode was a saturated calomel electrode, and the auxiliary electrode was made of platinum. The potentials of peak maxima were measured with V7-21 digital voltmeter with the precision ±1 mV. Triangular-shaped polarizing voltage was applied on the cell electrodes at a scanning rate of v=0.5 V/s. The potential was varied in the range from -0.2 to -0.4 V. The dissolved oxygen was removed from solutions by blowing purified argon over 20 min. pH values were measured with an ESL-43-07 glass electrode against an EVL-1M3.1 silver-silver chloride reference electrode using an I-160 M laboratory potentiometer (Belarus’).
Titration
The titrant volume was measured using a 10 mL microburette with the accuracy of ±0.01 mL.
Procedure
Kinetic-spectrophotometric method
0.35 g portion of the preparation powder was dissolved in twice-distilled water (100 mL). A 10.00 mL aliquot of the solution obtained, 2.0 mL of 0.02 M potassium caroate solution and 2.0 mL of 0.01 M of sodium hydroxide was transferred into 100 mL volumetric flask. The volume was brought to the mark and mixed. After the addition of NaOH a stop watch was switched on. Absorbance measurements at 305 nm were carried out every 2 min, during a total of 15 min after the preparation of the samples.
Voltammetric method
0.5000 g portion of cephalexin substance was dissolved in twice-distilled water (100 mL). A 5.5 mL portion of a 2.0 10-2 M of potassium caroate solution was added to 10 mL of the solution prepared, and the mixture was stirred carefully. A 5.0 mL portion of the solution prepared was transferred into a 50 mL volumetric flask, 5.0 mL of a 0.3 M acetate buffer solution with pH 4.0 was added, and the mixture was diluted to the mark with twice-distilled water and stirred carefully. The solution was charged into an electrolyser and voltammogram was recorded in the range from -0.2 to -1.4 V.
Iodometric method
0.36 g of cephalexin substance was dissolved in a in twice-distilled water (1000 mL). A 10.00 mL portion was transferred into 100 mL volumetric flask, 10.00 mL of 0.02 M KHSO5 solution was added and brought to the mark, stirred vigorously and left for 1 min. A 10.00 mL aliquot of the mixture obtained was transferred using the pipette into titration flask, 1 mL of 0.1 M sulfuric acid solution and 1 mL of 5 % potassium iodine solution were added. The isolated iodine was titrated by 0.02 M the titrated solution of sodium tiosulfate (V, mL). The control experiment was carried out in the same conditions paralleled (without Cephalexin with the same amount of KHSO5 0.02 M solution (V0, mL). Each one mL of 0.01 M solution of sodium tiosulphate is equivalent to 0.003654 g of cephalexin monohydrate (C16H17N3O4S) (content of cephalexin dehydrate in the limits 95-101%).
Method validation
The method was validated according to the guidelines of the International Conference on Harmonization17.
The Precision and Accuracy on these procedures were investigated with respect to repeatability and determined by performing five repeated analysis of the samples on the same day, under the same experimental conditions.
The Linearity was determined for a wide range of concentration for each of the methods. The calibration curves for each method were obtained, each research comprises 7 experimental points.
LOD and LOQ were calculated from regression equation as 3.3 S0/b and 10 S0/b respectively, where S0 and b are standard deviation slopes of the calibration curve.
Method comparison. Results obtained in this study were compared to those given in the quality certificates for the pure substance, i.e. HPLC method. The F-data comparison was performed between repeatability of the results obtained by iodometric method and by proposed kinetic-spectrophotomteric and voltammetric methods. The F-values should be less than 6.39.
RESULTS AND DISCUSSION
Kinetic-spectrophotometric method
In a basic medium Cephalexin S-oxide undergoes hydrolytic cleavage. Figure 1 shows the electronic spectra of Cephalexin and the reaction product. The appearance of a new band with absorption λmax = 305 nm demonstrates its formation in the reaction of alkaline hydrolysis of Cephalexin S-oxide in the presence of potassium caroate (perhydrolysis reaction).
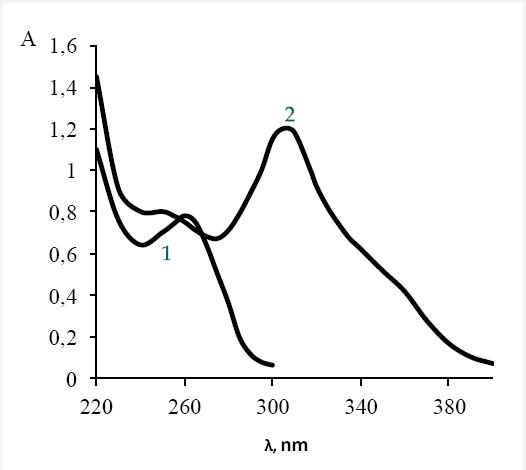
Fig. 1 Electronic spectra of Cephalexin (1) and S-reaction and perhydroolysis conjugated reaction with potassium caroate (2) absorbance. c(CF) = 1x10-4 M; c(KHSO5) = 2x10-4 M; c(NaOH) = 0.01 M
The interaction between Cephalexin and potassium caroate in acidic medium (pH = 3-5) is stoichiometric and fast: 1 mol of Cephalexin per 1 mol of KHSO5 (observation time is 1 min). In the basic medium a perhydrolysis conjugated reaction proceeds with formation of a corresponding product (Fig. 2, b).
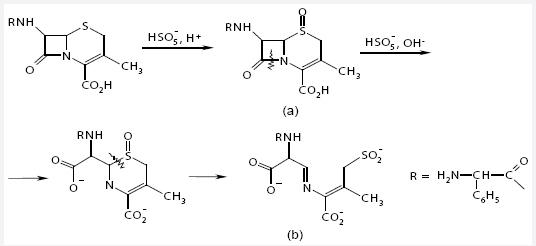
Fig. 2 The scheme of Cephalexin transformation: a) S-oxide product, b) S-reaction and perhydrolysis conjugated reaction with potassium caroate
During the experiment, it was determined that the order of mixing influences the kinetics and yield of the reaction. The highest rate of product accumulation was observed only after a prior addition of a potassium caroate solution to cephalosporin solution (formation of Cephalexin S-oxide), and then to a basic solution, optimal concentrations: potassium caroate 8 10-4 M, alkaline 2.1 10-2 M at 298-299 K. The reaction product wasn’t formed without oxidant during the first 30 min. The product formation rate was assessed by the slope (tgα, min-1) of linear plots of kinetic absorption curves, A versus time, t (in minutes).
Validation of the proposed method has been carried out for the parameters like linearity, precision, accuracy, LOD and LOQ. Detection wavelength selected for analysis was 305 nm. Linearity has been studied over a small drug concentration range from 1-50 µg mL-1. The correlation coefficient r=0.999 obtained for regression line showed good relationship between the tangent of the initial rate of the reaction and molar concentration of Cephalexin. The results of the proposed method are shown in the Tables 1-3.
Table 1 Experimental data obtained in the recovery test for Cephalexin pure substance by voltammetric (VM), kinetic-spectrophotometric (KSM) and iodometric (IM) methods
| Method | Added, g | Found, g | Recovery, % |
|---|---|---|---|
| VM | 0.3654 | 0.3428 | 98.70 |
| 0.2741 | 0.2719 | 99.20 | |
| 0.1827 | 0.1816 | 99.40 | |
| KSM | 0.3654 | 0.3661 | 100.2 |
| 0.2741 | 0.2766 | 100.92 | |
| 0.1827 | 0.1840 | 100.71 | |
| IM | 0.3654 | 0,3705 | 101.41 |
| 0.2741 | 0.2766 | 100.91 | |
| 0.1827 | 0,1823 | 99.79 |
Table 2 Analysis of Cephalexin pure substance accuracy determination
| Method | Mean, % | RSD, % | N | δ, % | F | |
|---|---|---|---|---|---|---|
| VM | 99.1 | 3.01 | 5 | -1.78 | 1.07 | F=<6.39 |
| KSM | 100.61 | 2.09 | 5 | -0.29 | 0.45 | |
| IM | 100.71 | 1.42 | 5 | -0.19 | * | |
*Reference method for F-data
Table 3 Characteristics of linear dependence for Cefalexin pure substance determination
| Method | Concentration range, μg mL-1 | Y = a±Sa + b±Sb ·X | R | LOD μg mL-1 | LOQ μg mL-1 |
|---|---|---|---|---|---|
| VM | 2-45 | I = (1.55 ± 0.1)104C + (0.04 ± 0.01) | 0.996 | 1.0 | 3.0 |
| KSM | 1-50 | tgα=(0,3621± 0,013)10-2 C | 0.999 | 0.3 | 1.0 |
| IM | 50-350 | Y = (1.025±0.13)X | 0.999 | 16 | 50 |
Voltammetry
Cephalexin S-oxide in the reaction under study is formed through an electrophilic attack of the β-oxygen atom in the peroxide group of the peroxoacid to sulfur within 1 min, i.e., the time of observation. The scheme of Cephalexin oxidation by potassium caroate to give the respective S-oxide is presented (Fig 2, a).
Three peaks were recorded in the cathodic branch of the voltammograms of solutions: at –0.485 V (average sharp), at –0.800 V (average smooth), and –1.120 V (small smooth, Fig. 3). Considerable changes in peak height at –0.800 V was observed with changes in cephalexin concentration. It was chosen for analytical purposes.

Fig. 3 An oscillographic polarogram of cephalexin S-oxide prepared by the reaction of Cephalexin with potasssium caroate.
The calibration plot was obtained. The relation of current I, µA, at –0.800 V to the concentration of cephalexin c, mol L-1, was approximated by the equation I = (1.55 ± 0.1) 104c + (0.04 ± 0.01), r = 0.996. The plot is linear in the range (1–10) 10–5 M. This made possible the further determination by a reference method.
The data of Cephalexin determination in the pure substance is given in the Tables 1-3.
Iodometry
The proposed method is based on the S-oxidation reaction of Cephalexin by potassium caroate in acidic medium. The oxidation-reduction interaction was determined to be quantitative and stoichiometric: 1 mol of KHSO5 per 1 mol of Cephalexin. The reaction product is cephalexin S-oxide (Fig.2, a).
The range of analytical procedure is the interval between 80% and 120% as recommended in SPhU.
Linearity is significant on the whole range of analyzed concentrations (0.05-0.35 mg mL-1). The linear equation generated from the calibration curve was Y = (1.025±0.13)X with a correlation coefficient r=0.9997. From this equation the content of cephalexin in the sample was calculated.
The detection limit (LOD) and the limit of quantification (LOQ) are less than 32% and do not influence on the quantitative determination.
The Limit of Quantitation (LOQ) is 0.05 mg mL-1.
The advantages of the proposed method are ability of analytical determination of Cephalexin by the biologically active part of the molecule, mainly alicyclic Sulphur, good precision and accuracy. The method validated is faster compared to standard iodometric procedure (approx. 40 min).
The results of Cephalexin iodometric determination are given in the Tables 1-3.
DETERMINATION OF CEPHALEXIN IN SUSPENSION
According to the instruction the suspension was prepared as followed: 74 mL of water was added into the bottle with granules and stirred carefully. The solution was filtrated, 5.00 mL of filtrate was transferred into a 100 mL volumetric flask and diluted to the mark with twice-distilled water. Furhter analysis was performed as in the procedures described for iodometric, kinetic-spectrophotometric and voltammetric methods. The results obtained are summarized in Table 4.
CONCLUSIONS
Potassium caroate is used as analytical reagent for Cephalexin.
The proposed procedures were developed and validated. The obtained results show good agreement with the standard HPLC (δ<RSD). The recovery percent ranged from 98.7 to 101.4. RSD from 1.42 to 3.0. The methods proposed are linear in a wide range: voltammetry 2-45 μg mL-1, kinetic-spectrophotometric method 1-50 μg mL-1, and iodometry 0.05-0.35 mg mL-1.
The iodometric method has the highest accuracy and precision RSD=1.42 %, δ=-0.19%.
The kinetic-spectrophotometric method is the most sensitive from the proposed (LOQ=1.0 μg mL-1), it has higher accuracy and precision if compared to the voltammetric method (F=0.45 <6.39).
All the developed procedures for the determination of Cephalexin in pure substance do not require elaborate treatment and expensive materials. The proposed methods are sensitive enough to enable determination of lower amounts of drug, these advantages encourage the application of proposed procedures in routine quality control of Cephalexin in industrial laboratories. Finally, these procedures provide advantages of improving selectivity, rapid, easy in performance and of a low cost.

