










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNefrología (Madrid)
On-line version ISSN 1989-2284Print version ISSN 0211-6995
Nefrología (Madr.) vol.35 n.1 Cantabria 2015
https://dx.doi.org/10.3265/Nefrologia.pre2014.Sep.12623
CASO CLÍNICO
Tumor de células yuxtaglomerulares como causa de hipertensión curable: presentación de un caso
Juxtaglomerular cell tumour as a curable cause of hypertension: case presentation
Jeremías T. Sierra1, Diego Rigo2, Agustín Arancibia3, Jorge Mukdsi4, Silvia Nicolai2 y M. Elvira Ortiz1
1Servicio de Medicina Interna. Hospital San Roque. Córdoba (Argentina)
2Servicio de Nefrología. Hospital San Roque. Córdoba (Argentina)
3Servicio de Patología. Hospital Privado. Córdoba (Argentina)
4Centro de Microscopía Electrónica. Facultad de Ciencias Médicas. Universidad Nacional de Córdoba. (Argentina)
Dirección para correspondencia
RESUMEN
La hipertensión arterial es una enfermedad de alta prevalencia y siempre deben tenerse en cuenta las causas secundarias que pueden originarla, debido a que el tratamiento y el pronóstico difieren entre estas y la hipertensión esencial. Presentamos el primer caso reportado en Argentina de una paciente de 21 años con hipertensión arterial e hipopotasemia, debido a un tumor de células yuxtaglomerulares productor de renina, el cual fue diagnosticado tras siete años de evolución.
Palabras clave: HTA secundaria, Hipopotasemia, Tumor de células yuxtaglomerulares.
ABSTRACT
Arterial hypertension is a highly prevalent disease and its secondary causes must always be kept in mind because the treatment and prognosis differ between these and essential hypertension. Here we present the first reported case in Argentina of a 21-year-old patient with arterial hypertension and hypokalaemia due to a renin-secreting juxtaglomerular cell tumour, which was diagnosed after seven years of development.
Key words: Secondary hypertension, Hypokalemia, Juxtaglomerular cell tumor.
Introducción
El reninoma es un tumor de las células del aparato yuxtaglomerular que produce cantidades excesivas de renina, resultando en una forma secundaria de hiperaldosteronismo, manifestándose clínicamente con hipertensión (HTA) e hipopotasemia1. Este tumor renal predomina en el sexo femenino, con un pico de incidencia entre la segunda y la tercera década de vida y, aunque infrecuente, es una causa tratable de HTA2. Debe sospecharse en cualquier paciente con HTA refractaria asociada a hipopotasemia y niveles elevados de aldosterona.
Presentamos el primer caso de reninoma reportado en Argentina en una mujer joven con HTA refractaria e hipopotasemia.
Caso clínico
Mujer, diagnosticada de HTA a los 14 años, cuando fue admitida por primera vez en un hospital pediátrico, con presión arterial (PA) de 210/120mmHg, cefaleas e hipopotasemia. Con la sospecha de HTA secundaria, se le realizaron diversos estudios. La ecografía renal, el ecocardiograma y el cateterismo de aorta y arterias renales no evidenciaron alteraciones. Los dosajes de actividad de renina plasmática (ARP) y aldosterona plasmática (AP) resultaron elevados en dos ocasiones (tabla 1). Los niveles de ácido vanilmandélico en orina y los de TSH, T4 libre y T3 plasmáticos resultaron normales. Recibió el alta hospitalaria medicada con inhibidores de la enzima convertidora de angiotensina y bloqueante cálcico, pero no regresó a control.
A los 19 años de edad, consultó en otro nosocomio por HTA y cursando embarazo de ocho semanas, por lo cual fue hospitalizada en terapia intensiva. Se determinó buena salud fetal, PA 200/110mmHg e hipopotasemia, iniciándose tratamiento con clonidina y alfa metil dopa. En ecografía renal se evidenció en polo inferior de riñón izquierdo formación nodular regular de 45mm de diámetro, ecográficamente compatible con angiomiolipoma. Los niveles de ARP y AP estaban elevados. Continuó en tratamiento ambulatorio con labetalol y nifedipina. A las 19 semanas de gestación fue admitida por guardia, con diagnóstico de aborto retenido, realizándose legrado uterino. Cuatro días después solicitó alta voluntaria, no regresando a control.
Dos años más tarde, estando medicada con enalapril, losartán y amlodipina, ingresa a la guardia de nuestro hospital consultando por cefalea occipital y debilidad muscular de varios días de evolución. Se constata PA de 220/140mmHg. En sus estudios iniciales se detectó hipokalemia de 2mEq/l, creatinina plásmatica de 0,6mg/dl y urea plasmática de 25mg/dl. La ecografía renal evidenció una imagen nodular sólida, corroborada por tomografía computarizada abdominal contrastada, donde se observó una voluminosa formación ocupante de espacio, sólida, en sector interpolar y polo inferior del riñón izquierdo de 65mm de diámetro, hipovascularizada (figura 1). El Doppler de arterias renales y el ecocardiograma no mostraron alteraciones.
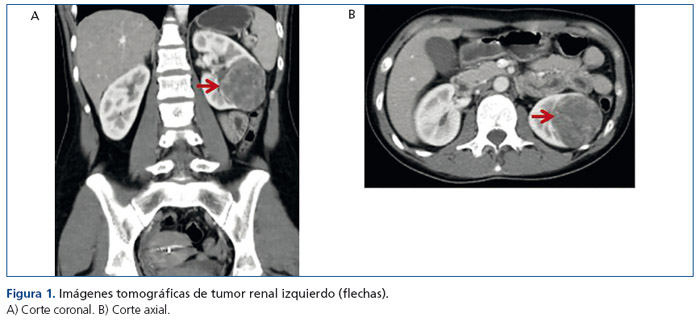
Ante el diagnóstico presuntivo de HTA secundaria a tumor renal, por los niveles elevados de renina, y habiéndose descartado otras causas secundarias de HTA en estudios anteriores, se decide realizar nefrectomía del riñón afectado, previamente controlada la PA con el agregado de espironolactona en dosis de 100mg/día a su medicación habitual.
El examen patológico reveló un tumor encapsulado de 5,5x4,5cm, de aspecto hemorrágico, con áreas fibrosas de color blanquecino. Microscópicamente la lesión tumoral se encontró constituida por células de mediano tamaño, con núcleos medianos, vesiculosos, con cromatina irregular, algunas con nucléolo inconspicuo y citoplasma eosinófilo, otras con citoplasma claro, dispuestas en un patrón difuso con extensa hemorragia y leve infiltrado inflamatorio mononuclear (figura 2 A y figura 2 B), sin invasión de la cápsula renal. No se encontró atipia celular, mitosis, ni necrosis. Las células tumorales fueron positivas para vimentina y mostraron una positividad fuerte y difusa para CD34 (figura 2 C). La observación a nivel ultraestructural permitió reconocer células con gránulos intracitoplasmáticos con diferentes grados de electrolucidez, rodeados por membranas. En cuanto a la morfología, variaron de redondos a alongados, adquiriendo algunas formas con bordes rectilíneos, y se destacó en algunos de ellos una forma romboidal cristaloide que a grandes magnificaciones presenta una estructura de zigzag de marcada electrodensidad (figura 2 D). Hallazgos vinculables a tumor de células yuxtaglomerulares (TCY), permitiendo corroborar la sospecha clínica.
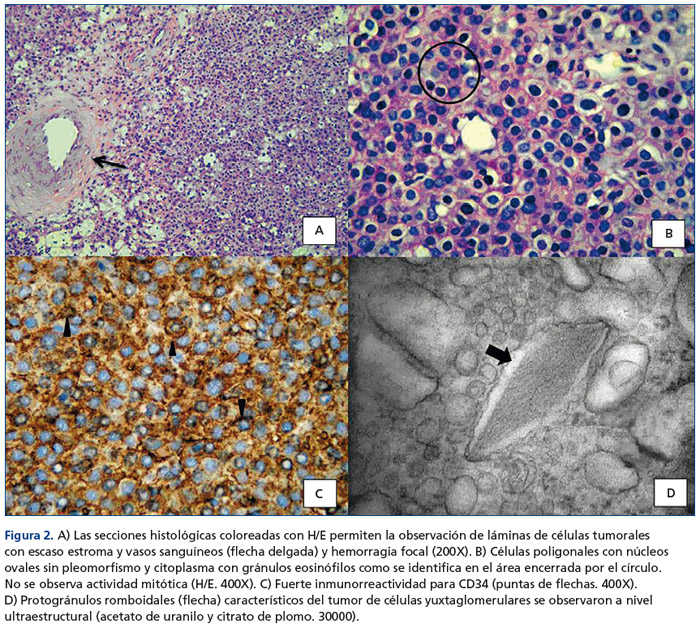
Posterior a la cirugía, la PA de la paciente fue disminuyendo de forma paulatina y simultáneamente se retiraron antihipertensivos, evolucionando asintomática, normotensa, con niveles de renina plasmática, aldosterona y potasio séricos normales, sin tratamiento farmacológico.
Discusión
La asociación de HTA e hipopotasemia orienta generalmente al diagnóstico de hiperaldosteronismo. Los valores elevados de ARP y de AP permiten el diagnóstico diferencial entre un hiperaldosteronismo primario y secundario3,4. Ante la presencia de un hiperaldosteronismo hiperreninémico e HTA, debe realizarse el diagnóstico diferencial entre estenosis de arterias renales, coartación de aorta y un tumor secretor de renina5.
En 1967, Robertson et al.6 describieron el primer tumor productor de renina. Kihara et al. fueron quienes posteriormente lo denominaron "tumor de células yuxtaglomerulares"7. Son tumores muy poco frecuentes, con un pico de incidencia entre la segunda y la tercera década de vida, y con predominancia por el sexo femenino, originados en células mioendocrinas del aparato yuxtaglomerular renal, aunque también se ha comprobado la producción de renina por otros tumores diferentes, como tumores de Wilms, tumores carcinoides, oncocitomas renales y carcinomas de células renales8,9. De acuerdo con la presencia o ausencia de síntomas, los TCY pueden presentarse como: una variante "típica", que representa la mayoría de los casos, y se manifiesta con HTA, hipokalemia, renina plasmática elevada e hiperaldosteronismo secundario; una variante "atípica", en la cual la HTA no se acompaña de hipokalemia; y una tercera variedad "no funcionante" que cursa sin HTA y con niveles normales de potasio10,11.
Los TCY se localizan en la corteza renal y en los primeros años de su aparición pueden ser indetectables por métodos imagenológicos comunes, por lo que se puede requerir la medición de renina de forma selectiva en cada vena renal para su diagnóstico12. El cateterismo de las arterias renales, además de descartar la presencia de estenosis en estas, puede detectar un área avascular correspondiente al sitio del tumor. La tomografía dinámica muestra un patrón típico que ayuda a diferenciar este tumor de otros, manifestándose como una masa hipovascular en la fase arterial, con captación moderada de contraste en la fase tardía. La resonancia magnética es una opción para tumores muy pequeños13.
El estudio anatomopatológico e inmunohistoquímico (inmuno-reactividad para los marcadores: CD34, vimentina, CK7 y citoqueratina CAM5; marcación variable para CD117 y actina del músculo liso; y negativa para factor de von Willebrand, CD31, desmina, proteína S-100, antígeno relacionado con melanosoma, cromogranina A, sinaptofisina y enolasa neuroespecífica), junto con la microscopía electrónica de transmisión, la cual revela la presencia de células con protogránulos y gránulos maduros de renina, permiten confirmar la sospecha clínica y realizar el diagnóstico diferencial con otros tumores, como el carcinoma de células renales, el hemangiopericitoma y el adenoma metanéfrico14. Si bien los TCY se consideran tumores de comportamiento benigno, la invasión vascular puede estar presente, como en el caso reportado por Shera et al., quienes demostraron invasión vascular, de la cápsula tumoral y de un ganglio linfático, con recidiva de la enfermedad al año15.
La genética molecular ha sido poco estudiada. La trisomía del cromosoma 10, la monosomía del cromosoma 9 y X, y la pérdida de la mayor parte del brazo del cromosoma 11q son los trastornos más detectados16.
La extirpación del tumor, con conductas quirúrgicas acordes al tamaño, logra la normalización de los niveles de potasio, renina y aldosterona, y en la mayoría de los casos también normaliza las cifras de PA17.
Habitualmente, esta clase de tumores se diagnostican tras 5-7 años desde el inicio de la HTA, por lo que ante la presencia de HTA e hipopotasemia deben estudiarse la ARP y la AP. Si ambas se hayan elevadas y el paciente no se encuentra bajo tratamiento con diuréticos, deben descartarse mediante eco-Doppler, angio-TAC o cateterismo arterial la coartación de aorta y la estenosis de arterias renales.
En conclusión, el TCY es una causa muy poco frecuente de HTA secundaria y existen reportados en la literatura poco más de 100 casos desde 1967. Este es el primero comunicado en nuestro país y se realizó tras varios años de evolución, ya que el tumor no se evidenció en un principio pese a los numerosos estudios, lo cual podría sugerir que las manifestaciones clínicas se inician con lesiones microscópicas. En estos casos, el dosaje selectivo de renina en cada vena renal sería una herramienta útil para diagnosticar el riñón afectado.
Agradecimientos
Apreciamos la colaboración en el seguimiento de la paciente durante la hospitalización al Dr. Eduardo Bravo, el Dr. Ricardo Pedernera y la Dra. Vanesa Perrone. Finalmente, agradecemos la asistencia técnica en la microscopía electrónica de transmisión de la Sra. Elena Pereyra.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Referencias Bibliográficas
1. Gottardo F, Cesari M, Morra A, Gardiman M, Fassina A, Dal Bianco M. A kidney tumor in an adolescent with severe hypertension and hipokalemia: an uncommon case-case report and review of the literature on reninoma. Urol Int 2010;85:121-4. [ Links ]
2. Wong L, Hsu T, Perlroth M, Hofmann L, Haynes C, Katznelson L. Reninoma: case report and literature review. J Hypertens 2008;26:368-73. [ Links ]
3. Kuroda N, Gotoda H, Ohe C, Mikami S, Inoue K, Nagashima Y, et al. Review of juxtaglomerular cell tumor with focus on pathobiological aspect. Diagn Pathol 2011;6:80. [ Links ]
4. Kallistratos MS, Giannakopoulos A, German V, Manolis AJ. Diagnostic modalities of the most common forms of secondary hypertension. Hellenic J Cardiol 2010;51:518-29. [ Links ]
5. Gupta V. Mineralocorticoid hypertension. Indian J Endocrinol Metab 2011;15(Suppl 4):s298-312. [ Links ]
6. Robertson PW, Klidjian A, Harding LK, Walters G, Lee MR, Robb-Smith AH. Hypertension due to a reninsecreting renal tumour. Am J Med 1967;43:963-76. [ Links ]
7. Kihara I, Kitamura S, Hoshino T, Seida H, Watanabe T. A hitherto unreported vascular tumor of the kidney: a proposal of "juxtaglomerular cell tumor". Acta Pathol Jpn 1968;18:197-206. [ Links ]
8. Lee SH, Kang MS, Lee GS, Chung WY. Refractory hypertension and isosexual pseudoprecocious puberty associated with renin-secreting ovarian steroid cell tumor in a girl. J Korean Med Sci 2011;26:836-8. [ Links ]
9. Tryfon S, Parisis V, Ioannis K, Saroglou M, Leonidas S, Despina D, et al. Excessive muscle paralysis due to pulmonary carcinoid-a case report. Clin Med Insights Case Rep 2012;5:43-8. [ Links ]
10. Dong D, Hanzhong L, Weigang Y, Weifeng X. Juxtaglomerular cell tumor of the kidney - a new classification scheme. Urol Oncol 2010;28:34-8. [ Links ]
11. Sakata R, Shimoyamada H, Yanagisawa M, Murakami T, Makiyama K, Nakaigawa N, et al. Nonfunctioning juxtaglomerular cell tumor. Case Rep Pathol 2013;2013:973865. [ Links ]
12. Wolley M, Gordon R, Stowasser M. Reninoma: the importance of renal vein rennin ratios for lateralisation and diagnosis. Am J Nephrol 2014;39:16-9. [ Links ]
13. Tanabe A, Naruse M, Ogawa T, Ito F, Takagi S, Takano K, et al. Dynamic computer tomography is useful in the differential diagnosis of juxtaglomeular cell tumor and renal cell carcinoma. Hypertens Res 2001;24:331-6. [ Links ]
14. Kim CH, Park YW, Ordonez NG, Ayala AG, Burroughs JF, Ro JY. Juxtaglomerular cell tumor of the kidney: case report with immunohistochemical and electron microscopic investigations and review of the literature. Int J Surg Pathol 1999;7:115-23. [ Links ]
15. Shera AH, Baba AA, Bakshi IH, Lone IA. Recurrent malignant juxtaglomerular cell tumor: A rare cause of malignant hypertension in a child. J Indian Assoc Pediatr Surg 2011;4:152-4. [ Links ]
16. Brandal P, Busund LT, Heim S. Chromosome abnormalities in juxtaglomerular cell tumors. Cancer 2005;104:504-10. [ Links ]
17. Haab F, Duclos JM, Guyenne T, Plouin PF, Corvol P. Renin secreting tumors: diagnosis, conservative surgical approach and long-term results. J Urol 1995;153:1781-4. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Jeremías T. Sierra
Servicio de Medicina Interna
Hospital San Roque
Córdoba, Argentina
jeremias.sierra@hospitalprivadosa.com
Enviado a Revisar: 10 Jun. 2014
Aceptado el: 16 Sep. 2014

