










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNefrología (Madrid)
On-line version ISSN 1989-2284Print version ISSN 0211-6995
Nefrología (Madr.) vol.35 n.3 Cantabria 2015
https://dx.doi.org/10.1016/j.nefro.2015.05.002
CASO CLÍNICO-CLUB DE NEFROPATOLOGIA
Amiloidosis renal hereditaria por depósito de apolipoproteína AI: un reto diagnóstico
Hereditary apolipoprotein AI-associated renal amyloidosis: A diagnostic challenge
Kelly del Rocío Samillán-Sosaa, Gloria Sención-Martíneza, Vanessa Lopes-Martína, Miguel Angel Martínez-Gonzálezb, Manel Soléc, Jose Luis Arosteguid, Jose Mesae, Juan de Dios García-Díaze, Diego Rodríguez-Puyola y Patricia Martínez-Miguela
a Servicio de Nefrología, Hospital Universitario Príncipe de Asturias, Alcalá de Henares, Madrid, España
b Servicio de Anatomía Patológica, Hospital Universitario 12 de Octubre, Madrid, España
c Servicio de Anatomía Patológica, Hospital Clínic, Barcelona, España
d Servicio de Inmunología, Hospital Clínic, Barcelona, España
e Unidad de Genética Clínica, Medicina Interna, Hospital Universitario Príncipe de Asturias, Alcalá de Henares, Madrid, España
Dirección para correspondencia
RESUMEN
La amiloidoisis renal hereditaria es un trastorno autosómico dominante cuya clínica se solapa con la de otros tipos de amiloidoisis. Hacer un adecuado diagnóstico diferencial puede ser difícil, pero tiene una gran relevancia respecto al pronóstico y tratamiento, que difiere según sea el origen de la enfermedad. Presentamos el caso clínico de un paciente con síndrome nefrótico e insuficiencia renal progresiva, con antecedente familiar de madre con amiloidosis renal.
En la biopsia renal se observó depósito de amiloide a nivel glomerular, con negatividad para cadenas ligeras y proteína AA. La sospecha clínica inicial fue la de amiloidosis por depósito de cadena A alfa de fibrinógeno, que es la causa más frecuente de amiloidosis hereditaria en Europa, con afectación preferentemente glomerular. Sin embargo, el estudio genético determinó una nueva mutación previamente no descrita de la Apolipoproteina AI (APO AI). En la biopsia de la madre se detectó depósito glomerular, pero también depósito masivo en médula, lo que caracteriza a la amiloidosis por depósito de APO AI. El diagnóstico se confirmó mediante inmunohistoquímica.
La amiloidosis por depósito de Apo AI progresa a enfermedad renal crónica terminal en el plazo de de 3 a 15 años. Se diferencia clínicamente de la amiloidosis AL por su menor afectación extrarrenal y su mejor pronóstico. El trasplante renal ofrece una supervivencia del injerto aceptable y el trasplante hepato-renal se podría tener en cuenta en pacientes con disfunción significativa de ambos órganos.
Palabras clave: Amiloidosis renal hereditaria. Apolipoproteína AI. Enfermedad renal progresiva.
ABSTRACT
Hereditary renal amyloidosis is an autosomal dominant condition with considerable overlap with other amyloidosis types. Differential diagnosis is complicated, but is relevant for prognosis and treatment. We describe a patient with nephrotic syndrome and progressive renal failure, who had a mother with renal amiloidosis.
Renal biopsy revealed amyloid deposits in glomerular space, with absence of light chains and protein AA. We suspected amyloidosis with fibrinogen A alpha chain deposits, which is the most frequent cause of hereditary amyloidosis in Europe, with a glomerular preferential affectation. However, the genetic study showed a novel mutation in apolipoprotein AI. On reviewing the biopsy of the patient's mother similar glomerular deposits were found, but there were significant deposits in the renal medulla as well, which is typical in APO AI amyloidosis. The diagnosis was confirmed by immunohistochemistry.
Apo AI amyloidosis is characterized by slowly progressive renal disease and end-stage renal disease occurs aproximately 3 to 15 years from initial diagnosis. Renal transplantation offers an acceptable graft survival and in these patients with hepatorenal involvement simultaneous liver and kidney transplantation could be considered.
Key words: Hereditary renal amiloidosis. Apolipoprotein AI. Progressive renal disease.
Caso clínico
Varón 40 años que acude a urgencias por la aparición de edemas. Presenta el antecedente familiar de madre con amiloidosis renal no filiada. El informe de la biopsia renal de la madre refiere resistencia al permanganato, lo que parece descartar amiloidosis AA, e induce a considerar, en el momento en el que se hizo el diagnóstico, la posibilidad de una amiloidosis AL.
El paciente no recibe ningún tratamiento farmacológico. A la exploración física destaca presión arterial de 135/75 mmHg y edemas bimaleolares, sin otros datos de interés. Las pruebas de laboratorio confirman la presencia de síndrome nefrótico (albúmina sérica de 2,5 g/dl, colesterolemia de 275 mg/dl y proteinuria de 6 g/24 h), sin alteraciones en el sedimento y filtrado glomerular normal. El espectro electroforético en sangre y orina, el estudio inmunológico y la ecografía renal son normales. Se realiza biopsia renal, observándose 19 glomérulos con una estructura totalmente distorsionada por un depósito hialino en disposición nodular, con positividad para rojo Congo y tioflavina; los túbulos y los vasos no muestran alteraciones significativas, siendo el resultado compatible con amiloidosis. En el estudio sobre muestra en parafina se evidencia marcada positividad para proteína AP, que es una parte de la sustancia amiloide común en todos los tipos de amiloidosis, con negatividad para cadenas ligeras y proteínas AA, B2 microglobulina y transtirretina. Se realiza estudio complementario con anticuerpos frente a fibrinógeno y lisozima, resultando los depósitos de sustancia amiloide positivos para la cadena A alfa del fibrinógeno (Afib). Sin embargo, en el estudio genético no se encuentra ninguna mutación en el gen de la cadena Afib, y sí se demuestra una mutación previamente no descrita en el gen de la apolipoproteína AI (apo AI). Ante esta discrepancia se analiza la biopsia de la madre, que contiene depósito glomerular en la corteza y depósito masivo a nivel medular. El depósito a nivel medular no está descrito en la amiloidosis Afib, siendo muy característico de la apo AI. Se concluye que los primeros resultados de la inmunohistoquímica no son fiables por no haberse hecho en las condiciones adecuadas, confirmándose posteriormente mediante la misma técnica la presencia de apo AI.
Se realiza estudio de extensión, con ecocardiograma y electromiograma, sin evidencia de afectación cardiaca ni neuropatía autonómica. En la evolución se detecta un aumento progresivo de transaminasas, sugerente de afectación hepática. Estos resultados son concordantes con este tipo de amiloidosis familiar, con afectación preferentemente renal y hepática. A pesar de la normalidad inicial, la función renal se deteriora progresivamente hasta requerir terapia renal sustitutiva en un plazo de 2 años. El paciente recibe tratamiento con diálisis peritoneal durante 4 años, hasta que es intervenido realizándose trasplante renal de donante cadáver. Presenta un postoperatorio complicado con rotura esplénica, en el contexto de su enfermedad, con depósito de amiloide en bazo, siendo necesaria una esplenectomía urgente. Finalmente, el paciente es dado de alta en situación estable (figs. 1-4).
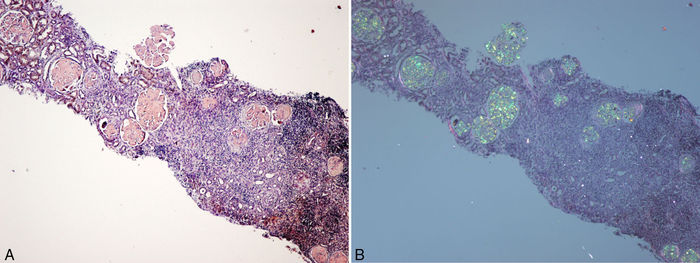
Figura 1 - A) Biopsia renal (hijo). Parénquima cortical con depósito glomerular masivo de
sustancia amiloide teñida con rojo Congo. B) Parénquima cortical con depósito glomerular
masivo de sustancia amiloide observada con luz polarizada. Destaca también la fibrosis y la
inflamación intersticial (×40).
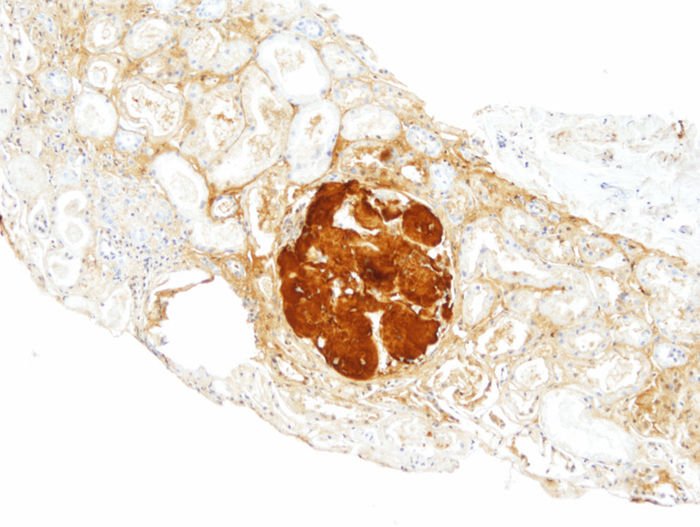
Figura 2 - Biopsia renal (hijo). Inmunohistoquímica para apo AI, con positividad intensa
en los depósitos glomerulares (×200).
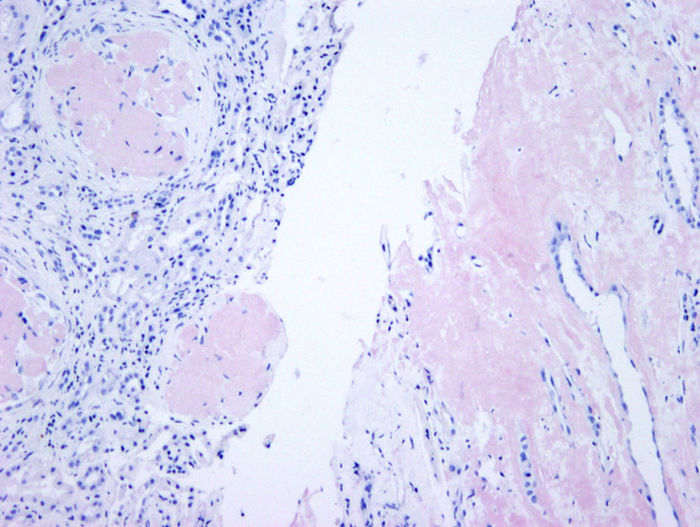
Figura 3 - Biopsia renal (madre). Depósito masivo glomerular en córtex (izquierda) e
intersticial en médula (derecha) (rojo Congo ×100).
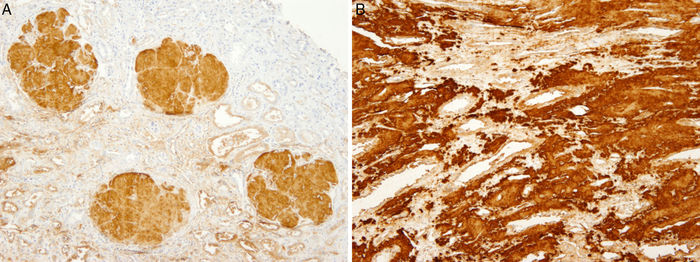
Figura - 4 A) Biopsia renal (madre). Inmunohistoquímica para apo AI positiva en los depósitos
glomerulares. B) Inmunohistoquímica para apo A1 positiva en los depósitos intersticiales de
médula renal.
Discusión
La amiloidosis renal hereditaria no neuropática fue descrita por primera vez por Ostertag en 19321. Se denominó así enfatizando la ausencia de afectación del sistema nervioso periférico, con el fin de diferenciar esta patología de otros tipos de amiloidosis. Sin embargo, este término es confuso y no apropiado en la actualidad, ya que posteriormente se han descrito algunas variantes relacionadas con la apo AI asociadas a neuropatía2. En relación con esta enfermedad se han identificado más de 25 mutaciones que alteran la estructura de diferentes proteínas, provocando su depósito en forma de amiloide. Estas proteínas son la lisozima3, la apo AI4,5, la apolipoproteína AII (apo AII)6,7 y la cadena Afib8-12. Clínicamente, todas ellas se manifiestan con insuficiencia renal en la edad media de la vida y tienen una herencia autosómica dominante, aunque con una penetrancia muy variable y unas características clínicas específicas, según sea la proteína que origina el depósito.
La apo AI es la principal proteína que conforma la estructura de las lipoproteínas de alta densidad (HDL). Es secretada en el hígado y en el intestino delgado, y se cataboliza fundamentalmente en el hígado y los riñones13-15. Es un cofactor para la lecitina colesterol acetil-transferasa (LCAT), y su función está vinculada a la eliminación del colesterol13,15,16.
Hasta la fecha se han descrito al menos 19 mutaciones amiloidogénicas del gen de la apo AI. Se ha podido observar una presentación clínica diferente de la enfermedad dependiendo de la región en la que se localice la mutación. De este modo, las mutaciones que afectan a la región aminoterminal se manifiestan con depósito de amiloide a nivel hepático y renal, mientras que las mutaciones que afectan a la región carboxiterminal se manifiestan con amiloidosis cardíaca, laríngea y cutánea2,15,17.
El fenotipo de la amiloidosis apo AI es muy heterogéneo. Se han publicado casos con una amiloidosis visceral extensa e insuficiencia renal terminal, mientras que en otros se ha detectado afectación de piel y laringe, con consecuencias clínicas mínimas. La expresividad clínica no solo depende de la ubicación de la mutación, ya que incluso pacientes con una misma mutación pueden tener una expresividad clínica diferente. La heterogeneidad fenotípica también se ha observado entre linajes con la misma mutación apo AI. Se han publicado series de pacientes con un elevado número de familiares afectados y alta progresión a enfermedad renal terminal (10 de 16 casos afectados)15, lo que sugiere la existencia de un fenotipo más agresivo u otros factores genéticos y ambientales que interactúan o influyen en las manifestaciones clínicas. El posible depósito de amiloide se ha descrito en múltiples órganos, entre ellos riñón, tracto gastrointestinal, bazo, hígado; corazón, sistema nervioso periférico, laringe y piel. La presentación clínica más habitual consiste en hipertensión arterial, proteinuria leve e insuficiencia renal lentamente progresiva entre los 18 y 55 años de edad2.
La primera mutación descrita, y la más común en pacientes con amiloidosis apo AI de ascendencia irlandesa, es la Gly26Arg. Se caracteriza por la progresiva acumulación de fibrillas de amiloide compuestas por los fragmentos de polipéptido aminoterminal de la apo AI, y se presenta en pacientes jóvenes, de entre 20 y 46 años de edad. El depósito de amiloide se localiza en los nervios periféricos, riñones, hígado y tracto gastrointestinal2,17, manifestándose clínicamente con neuropatía periférica, úlcera péptica y nefropatía2,18.
Posteriormente se han descrito al menos hasta 19 mutaciones amiloidogénicas de la apo AI. Nosotros describimos una nueva mutación que no hemos encontrado en la literatura, la c.220T>C (p.Trp74Arg). En el caso de nuestro paciente, pudimos observar afectación renal, con deterioro de la función renal en 2 años, así como afectación en hígado y bazo.
El análisis histológico en la amiloidosis apo AI muestra depósitos de amiloide predominante a nivel medular con un patrón de nefritis tubulointersticial, caracterizada por atrofia tubular y fibrosis intersticial, asociado a glomeruloesclerosis focal secundaria15,19. Aunque es predominante la afectación intersticial, también hay casos en los que se ha detectado depósito a nivel glomerular19-21. Incluso se ha publicado un caso con afectación predominante glomerular, asociado a síndrome nefrótico, como resultado de amiloidosis apo AI Leu64Pro22.
La preferente selectividad de la amiloidosis apo AI por la médula renal ayuda a diferenciarla de otros tipos de amiloidosis sistémicas hereditarias y adquiridas19,23, ya que únicamente comparte preferencia por el compartimento medular con la amiloidosis por depósito de transtirretina19,24.
En nuestro caso, la biopsia renal del paciente contenía solo corteza, y únicamente se localizó depósito amiloide en el compartimento glomerular. Estos hallazgos son concordantes con la clínica al diagnóstico, consistente síndrome nefrótico, con posterior empeoramiento progresivo de la función renal. Esta peculiaridad hizo sospechar inicialmente una amiloidosis por depósito de la cadena A alfa del fibrinógeno, ya que es la causa más frecuente de amiloidosis renal hereditaria en Europa, y su depósito es característicamente glomerular. Sin embargo, la biopsia de la madre tenía depósito glomerular en corteza, pero también contenía médula con depósito masivo. Finalmente, el estudio genético determinó una nueva mutación previamente no descrita de apo AI, y la inmunohistoquímica confirmó el diagnóstico.
La importancia de un correcto diagnóstico diferencial en la amiloidosis sistémica radica en que el manejo y el pronóstico de la enfermedad puede ser completamente diferente según sea el origen de la misma, para lo cual tiene especial relevancia determinar la proteína que origina el depósito. Según un estudio realizado en el Centro Nacional de Amiloidosis del Reino Unido, casi un 10% de los pacientes con un diagnóstico presuntivo de amiloidosis sistémica AL realmente padecen una amiloidosis hereditaria25. Esto se debe a que no es infrecuente considerar la amiloidosis AL como un diagnóstico de exclusión, debido al solapamiento clínico entre los diferentes tipos de amiloidosis, al relativamente frecuente hallazgo de una proteína monoclonal en pacientes mayores de 50 años, y a la dificultad para determinar el tipo de fibrilla, incluso siendo acertado el diagnóstico de amiloidosis AL, ya que hasta en un 20% de los casos los anticuerpos frente a las cadenas ligeras kappa o lambda no se unen a la misma, probablemente por la alteración en su estructura. Por otro lado, no siempre es posible constatar la presencia de antecedentes familiares en la amiloidosis hereditaria, debido a la variable penetrancia de la enfermedad y al diagnóstico de nuevas mutaciones.
Entre los métodos disponibles para determinar la composición proteica de las fibrillas se encuentran la inmunohistoquímica y la secuenciación directa de la proteína. Para realizar esta última técnica se requiere una cantidad considerable de tejido, algo que no siempre es posible obtener. El reciente desarrollo de técnicas de proteómica constituye un avance para el diagnóstico de la amiloidosis, ya que para identificar la proteína que conforma la fibrilla amiloidea se precisa únicamente una pequeña cantidad de tejido fijado26.
Respecto al tratamiento de la enfermedad, el trasplante renal en la amiloidosis apo AI ha tenido un éxito variable19,27,28. Se ha observado recidiva de la amiloidosis en el injerto, pero en la mayoría de los casos publicados la supervivencia del injerto es aceptable. Un estudio realizado en el Centro Nacional de Amiloidosis del Reino Unido reveló que de 10 receptores de trasplante renal con amiloidosis apo AI, 5 mostraron evidencia documentada del depósito de amiloide en el riñón trasplantado. Sin embargo, solo un paciente, después de 25 años, progresó a enfermedad renal terminal, iniciando nuevamente diálisis21,29.
El trasplante de hígado es el tratamiento definitivo en otros tipos de amiloidosis hereditaria, ya que en el hígado se sintetiza la proteína amiloidogénica. Sin embargo, en la amiloidosis por depósito de apo AI la proteína amiloidogénica también se sintetiza en el intestino delgado. Aunque se han descrito casos como el de un paciente irlandés con la mutación apo AI Gly26Arg, en el que se realizó un trasplante hepatorrenal y 2 años después se evidenció una regresión de los depósitos de amiloide19,30, el doble trasplante hepatorrenal solo se considera en pacientes con una disfunción significativa de ambos órganos.
En el caso de nuestro paciente, consideramos que la mejor opción era el trasplante renal, ya que la funcionalidad hepática no estaba muy deteriorada. Sin embargo, la evolución en el periodo postrasplante está siendo complicada, debido a rotura esplénica por depósito de amiloide.
Podríamos concluir que la amiloidosis renal hereditaria constituye un reto diagnóstico, dada la dificultad que existe para identificar la proteína que origina el depósito, y la importancia que tiene respecto al pronóstico y al tratamiento. En la amiloidosis por depósito de apo AI el diagnóstico de los casos índice requiere una alta sospecha clínica, y destaca la importancia de la biopsia de la médula renal para observar el depósito de amiloide a este nivel. Debe considerarse la posibilidad de este diagnóstico en casos familiares de nefritis tubulointersticial con afectación hepática.
Preguntas
1. Dra. Esther Rosello (Valencia). La rentabilidad diagnóstica para amiloidosis obliga muchas veces a realizar estudios en grasa, mucosa rectal y, al final, biopsia renal. ¿Qué decisión seguís en la obtención de biopsias ante sospecha de amiloidosis?
Respuesta (Kelly Samillán). Ante la sospecha de amiloidosis, el diagnóstico frecuentemente puede ser confirmado con la aspiración de grasa abdominal, que es una técnica poco invasiva. En la literatura, se refiere que esta técnica proporciona una alta sensibilidad para el diagnóstico de amiloidosis AL (entre el 80 y el 90%) y una sensibilidad más baja (entre el 65 y el 75%) para diagnosticar amiloidosis AA. Sin embargo en el caso de amiloidosis familiares la rentabilidad diagnóstica es muy baja y la ausencia de positividad al rojo Congo en una biopsia de grasa abdominal no excluye el diagnóstico31. En cuanto a las biopsias de glándulas salivales y de mucosa rectal, también son usadas como métodos diagnósticos no invasivos. Creemos que el método diagnóstico en los casos de amiloidosis debe individualizarse, según las características del paciente y su comorbilidad. Generalmente, si el paciente presenta una clara afectación renal, especialmente si tiene síndrome nefrótico, nuestro grupo indica la realización de biopsia renal, salvo casos excepcionales en los que la biopsia renal podría no ser determinante para tomar decisiones o en los que la comorbilidad del paciente no lo permita. En los casos de amiloidosis por depósito de apo AI con afectación renal, en nuestra opinión, es recomendable hacer directamente la biopsia renal.
2. Dra. Esther Roselló (Valencia). ¿Piensas que se podrían enviar a centros de referencia muestras de biopsias para técnicas especiales en amiloidosis?
Respuesta (Manuel Solé). Las dificultades en el diagnóstico de la amiloidosis se presentan a 2 niveles: el básico, el diagnóstico con la tinción de rojo Congo, debería ser resuelto en el propio centro, mediante el control adecuado de la técnica, aunque somos conscientes de los muchos problemas que presenta. Si se trata de tipificación, es importante utilizar un panel de anticuerpos y la interpretación requiere experiencia; en este caso, referir la muestra a una unidad de amiloidosis es adecuado.
3. Dr. Miguel Ángel Martínez (Madrid). Me gustaría comentar que las técnicas de rojo Congo solo salen bien si los cortes tienen un mínimo de 6 micras. ¿Coincide con vuestra experiencia?
Respuesta (Manuel Solé). Este es un aspecto técnico muy importante. Se suele aconsejar hacer la tinción sobre cortes de 8 micras. Sin embargo, no siempre es posible, especialmente en biopsias renales en las que hacen de rutina cortes sin teñir para diferentes técnicas. Si el corte es fino, la tinción será pálida, pero la birrefringencia verde se mantendrá, aunque pueda ser algo más difícil de visualizar.
4. Dra. Julia Blanco (Madrid). He notado que habéis conseguido en las técnicas de rojo Congo un verde manzana excelente. ¿Tenéis alguna fórmula secreta?
Respuesta (Manuel Solé). Es fundamental que la observación con luz polarizada se haga con una fuente de luz potente. No sirve cualquier microscopio. Y recordar que lo característico del rojo Congo es el dicroísmo, es decir, el paso de rojo a dorado y verde al rotar el filtro de polarización.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. Ostertag B. Demonstration einer eigenartigen familiaren para amyloidose. Zentralbl Aug Pathol. 1932;56:253-4. [ Links ]
2. Rowczenio D., Dogan A., Theis J.D., Vrana J.A., Lachmann H.J., Wechalekar A.D., et al. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am J Pathol.. 2011;179:1978-87. [ Links ]
3. Pepys M.B., Hawkins P.N., Booth D.R., Vigushin D.M., Tennent G.A., Soutar A.K., et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362:553-7. [ Links ]
4. Nichols W.C., Dwulet F.E., Liepnieks J., Benson M.D. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem Biophys Res Commun. 1988;156:762-8. [ Links ]
5. Obici L., Palladini G., Giorgetti S., Bellotti V., Gregorini G., Arbustini E., et al. Liver biopsy discloses a new apolipoprotein A-I hereditary amyloidosis in several unrelated Italian families. Gastroenterology.. 2004;126:1416-22. [ Links ]
6. Benson M.D., Liepnieks J.J., Yazaki M., Yamashita T., Hamidi Asl K., Guenther B., et al. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics. 2001;72:272-7. [ Links ]
7. Yazaki M., Liepnieks J.J., Yamashita T., Guenther B., Skinner M., Benson M.D. Renal amiloidosis caused by a novel stop-codon mutation in the apolipoprotein A-II gene. Kidney Int.. 2001;60:1658-65. [ Links ]
8. Benson M.D., Liepnieks J., Uemichi T., Wheeler G., Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen alpha-chain. Nat Genet. 1993;3:252-5. [ Links ]
9. Uemichi T., Liepnieks J.J., Benson M.D. Hereditary renal amyloidosis with a novel variant fibrinogen. J Clin Invest. 1994;93:731-6. [ Links ]
10. Uemichi T., Liepnieks J.J., Yamada T., Gertz M.A., Bang N., Benson M.D. A frame shift mutation in the fibrinogen A alpha chain gene in a kindred with renal amyloidosis. Blood. 1996;87:4197-203. [ Links ]
11. Hamidi Asl L., Liepnieks J.J., Uemichi T., Rebibou J.M., Justrabo E., Droz D., et al. Renal amyloidosis with a frame shift mutation in fibrinogen A alpha-chain gene producing a novelamyloid protein. Blood. 1997;90:4799-805. [ Links ]
12. Stangou A.J., Banner N.R., Hendry B.M., Rela M., Portmann B., Wendon J., et al. Hereditary fibrinogen A alfa-chain amyloidosis: Phenotypic characterization of a systemic disease and the role of liver transplantation. Blood. 2010;115:2998-3007. [ Links ]
13. Joy T., Wang J., Hahn A., Hegele R.A. APO A1 related amyloidosis: A case report and literature review. Clin Biochem. 2003;36:641-5. [ Links ]
14. Assman G., Shmitz G., Funke H., von Eckardstein A. Apolipoprotein A-I and HDL deficiency. Curr Opin Lipidol. 1990;1:110-5. [ Links ]
15. Traynor C.A., Tighe D., O'Brien F.J., Leavey S., Dorman A.M., Denton M.D., et al. Clinical and pathologic characteristics of hereditary apolipoprotein A-I amyloidosis in Ireland. 2013;18:549-54. [ Links ]
16. Sorci-Thomas M.G., Thomas M.J. The effects of altereted apolipoproteín A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121-8. [ Links ]
17. Eriksson M., Schonland S., Yumlu S., Hegenbart U., von Hutten H., Gioeva Z., et al. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: Identification of three novel mutations in the APOA1 gene. J Mol Diagn. 2009;11:257-62. [ Links ]
18. Van Allen M.W., Frohlich J.A., Davis J.R. Inherited predisposition to generalized amyloidosis. Clinical and pathological study of a family with neuropathy, nephropathy, and peptic ulcer. Neurology. 1969;19:10-25. [ Links ]
19. Gregorini G., Izzi C., Obici L., Tardanico R., Röcken C., Viola B.F., et al. Renal apolipoprotein A-I amyloidosis: A rare and usually ignored cause of hereditary tubulointerstitial nephritis. J Am Soc Nephrol. 2005;16:3680-6. [ Links ]
20. Vigushin D., Gough J., Allan D., Alguacil A., Penner B., Pettigrew N., et al. Familial nephropathic systemic amyloidosis caused by apolipoprotein AI variant Arg26. Q J M ed. 1994;87:149-54. [ Links ]
21. Booth D., Tan S., Booth S., Tennent G., Hutchinson W., Hsuan J., et al. Hereditary hepatic and systemic amyloidosis caused by a new deletion/insertion mutation in the apolipoprotein AI gene. J Clin Invest. 1996;97:2714-21. [ Links ]
22. Murphy C., Wang S., Weaver K., Gertz M., Weiss D., Solomon A. Renal apolipoprotein A-I amyloidosis associated with novel mutant Leu64Pro. Am J Kidney Dis. 2004;44:1103-9. [ Links ]
23. Benson M.D. The hereditary amyloidosis. Best Pract Res Clin Rheumatol. 2003;17:909-27. [ Links ]
24. Lobato L., Beirão I., Guimarães S.M., Droz D., Guimarães S., Grünfeld J.P., et al. Familial amyloid polyneuropathy type I (Portuguese): Distribution and characterization of renal amyloid deposits. Am J Kidney Dis. 1998;31:940-6. [ Links ]
25. Lachmann H.J., Booth D.R., Booth S.E., Bybee A., Gilbertson J.A., Gillmore J.D., et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-91. [ Links ]
26. Vrana J.A., Gamez J.D., Madden B.J., Theis J.D., Berger H.R., Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic análisis in clinical biopsy specimens. Blood. 2009;114:4957-9. [ Links ]
27. Falk R.H., Comenzo R.L., Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337:898-909. [ Links ]
28. Persey M.R., Booth D.R., Booth S.E., van Zyl-Smit R., Adams B.K., Fattaar A.B., et al. Hereditary nephropathic systemic amyloidosis caused by a novel variant apolipoprotein A-I. Kidney Int. 1998;53:276-81. [ Links ]
29. Gillmore J.D., Stangou A.J., Lachmann H.J., Goodman H.J., Wechalekar A.D., Acheson J., et al. Organ transplantation in hereditary apolipoprotein AI amyloidosis. Am J Transplant. 2006;6:2342-7. [ Links ]
30. Gillmore J.D., Stangou A.J., Tennent G.A., Booth D.R., O'Grady J., Rela M., et al. Clinical and biochemical outcome of hepatorenal transplantation for hereditary systemic amyloidosis associated with apolipoprotein AI Gly26Arg. Transplantation. 2001;71:986-92. [ Links ]
31. Dember M.L. Amyloidosis-associated kidney disease. J Am Soc Nephrol. 2006;17:3458-71. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Correo electrónico: pmmiguel@salud.madrid.org
(P. Martínez-Miguel).
Recibido el 30 de octubre de 2014
Aceptado el 5 de marzo de 2015

