










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkEFECTOS DEL EVEROLIMUS SOBRE LOS ANGIOMIOLIPOMAS RENALES SECUNDARIOS A ESCLEROSIS TUBEROSA
NR. ROBLES1, R. TORRA2, R. PECES3, J. NIETO4
1NEFROLOGÍA. HOSP INFANTA CRISTINA (BADAJOZ), 2NEFROLOGÍA. FUNDACION PUIGVERT (BARCELONA), 3NEFROLOGÍA. HOSP. LA PAZ (MADRID), 4NEFROLOGÍA. HOSP UNIVERSITARIO DE CIUDAD REAL (CIUDAD REAL)
La comunicación corresponde a un grupo de trabajo o un estudio multicentrico:
En nombre de los autores del ESTUDIO CRAD001MES12
Introducción: El complejo de la sclerosis tuberosa (CET) es una enfermedad genética causada por mutaciones que activan la via mTOR y llevan al crecimiento de tumores como angiomiolipomas (AML) renales. Sirolimus, un inhibidor de mTOR, ha demostrado ser una alternativa al tratamiento quirúrgico de los AML. Se ha evaluado el efecto de everolimus en un grupo de pacientes con AML secundarios a CET.
Diseño y Métodos: Ensayo clinico abierto de acceso expandido en fase III. Se incluyeron pacientes mayores de 18 años con AML renales secundarios a CET mayores de 3 cm de diametro mayor. Los pacientes fueron tratados con 10 mg/dia de everolimus pudiendo ajustarse las dosis en función de la evolución clínica. Se realizaron estudios de imagen (TC o RM) al inicio del estudio y cada 3 meses hasta completar un año de seguimiento.
Resultados: Se reclutaron 19 pacientes (mediana de edad, 38,0 [29,0-43,0] años; mujeres 68.4%). La mediana del volumen de las lesiones objetivo fue 260.0 [127,8-322,2] ml (riñon derecho) y 275,5 [173,4-402,4] ml (RI). Reducciones del volumen superiores al 30% fueron observadas en 11 (57,9%) pacientes y superaron el 50% 9 (47,4%) pacientes. Nueve pacientes (47,4%) mostraron respuesta radiológica, diez (52,6%) estabilidad y en ningún caso se detectó progresión. Once pacientes recibieron (57,9%) recibieron la dosis de 10 mg durante todo el estudio y 8 (42,1%) necesitaron reducción o interrupción temporal de la medicación. No hubo ningún caso de suspensión definitiva del tratamiento. Los efectos secundarios más frecuentes (>25% de los pacientes) fueron: estomatitis aftosa, hipercolesterolemia, hipertrigliceridemia, infecciones de tracto urinario, mucositis, hipertensión arterial, dermatitis acneiforme e insomnio. Solo un efecto adverso grave (neumonía) fue comunicado.
Conclusiones: El estudio confirma la eficacia del everolimus para reducir el tamaño de los angiomiolipomas secundarios a CET y el tamaño renal global, así como la seguridad de su uso en estos pacientes.
TRASPLANTE RENAL EN PACIENTES CON POLIQUISTOSIS RENAL: SEGUIMIENTO A LARGO PLAZO E IMPACTO DEL ANEURISMA CEREBRAL
C. BURBALLA TÀRREGA1, MJ. PÉREZ SÁEZ1, D. REDONDO PACHÓN1, M. MIR FONTANA1, L. SANS ATXER1, A. CALIFANO1, A. BUXEDA PORRAS1, AM. GRANADOS MARTINEZ1, M. CRESPO BARRIO1, J. PASCUAL SANTOS1
1NEFROLOGÍA. HOSPITAL DEL MAR (BARCELONA)
Introducción: La poliquistosis renal (PQR) representa el 7% de los pacientes que inician terapia renal sustitutiva en España; este porcentaje es mayor entre los pacientes prevalentes trasplantados renales (TR). Analizamos los resultados a largo plazo del TR en una cohorte de pacientes con PQR, en especial la prevalencia de aneurismas cerebrales y mortalidad asociada.
Métodos: Estudio retrospectivo TR 1979-2013. El examen con imagen cerebral de la presencia de aneurismas se ha realizado ante la presencia de síntomas o eventos, no como cribado en asintomáticos.
Resultados: De 709 pacientes, 98(13.8%) presentaban PQR como nefropatía de base. Los pacientes con PQR eran mayores (53,5±10,8 vs 47,8±14,6; p<0.0001) y en mayor número mujeres (50 vs 37.3%; p=0.004) que los no-PQR. La supervivencia del injerto censurada por muerte del paciente a 5 años fue mayor en los pacientes con PQR que no-PQR (72.9 vs 61.3%; p=0.04). A 10 años el tamaño muestral disminuye y se pierde la diferencia. La supervivencia del paciente a 3-5 años es mayor en PQR (Fig 1). El análisis multivariante no mostró asociación entre PQR y supervivencia del paciente/injerto.

El 42.8% (n=42) de los TR-PQR tenían prueba de imagen cerebral pre-TR, hallándose aneurismas en el 23.8% (n=10); de ellos, en 8 se decidió actitud conservadora y en 2 se realizaron clipajes. De los 32 sin hallazgos, dos fallecieron de hemorragia cerebral. Quince de los 98(15%) fallecieron durante la evolución; tres (20%) lo hicieron por rotura de aneurisma, 8 neoplasia, 3 cardiovascular y uno desconocida. De los 3 fallecidos por rotura, dos tenían TAC normal y el otro no tenía exploración.
Conclusiones: Los pacientes con PQR presentan mayor supervivencia del paciente e injerto a 3-5 años que los no-PQR. La hemorragia intracraneal por rotura de aneurisma es una causa importante de muerte en trasplantados con PQR. El cribado de aneurismas en pacientes asintomáticos podría estar indicado.
TRANSICION COORDINADA DEL PACIENTE CON CISTINOSIS DESDE LA MEDICINA PEDIATRICA A LA MEDICINA DEL ADULTO
G. ARICETA1, JA. CAMACHO2, E. LARA1, JV. TORREGROSA3, G. PINTOS-MORELL4, A. VILA-SANTAN-DREU5, N. MARTÍN-BEGUÉ6, S. TORRES1
1NEFROLOGÍA INFANTIL. H VALLE HEBRON (BARCELONA), 2NEFROLOGÍA INFANTIL. H SAN JOAN DE DEU (BARCELONA), 3NEFROLOGÍA. H CLINIC (BARCELONA), 4PEDIATRIA. H CAN RUTI (BADALONA), 5NEFROLOGÍA INFANTIL. H SANT JOAN DE DEU (BARCELONA), 6OFTALMOLOGIA INFANTIL. H VALLE HEBRON (BARCELONA)
Introducción: El aumento de la supervivencia de los pacientes con cistinosis y la complejidad de la enfermedad per se, explica la necesidad de implementar un proceso de transición guiada desde la medicina pediátrica a la del adulto, que permita garantizar el continuum asistencial y posibilite el empoderamiento del paciente desde el cuidado tutelado al autocuidado.
Métodos: El trabajo que presentamos es el resultado de diferentes estrategias metodológicas que llevó a cabo el Grupo T-CiS.bcn: 1) búsqueda bibliográfica en PubMed con las palabras clave “cistinosis”, “transición”, “adolescente”, “trasplante renal”; 2) encuesta directa a los propios integrantes del grupo T-CiS.bcn para identificar as necesidades y los retos de la transición en el propio entorno sanitario y favorecer la implementación de estrategias de mejora; 3) contacto con el Grupocistinosis que representa al grupo de pacientes y familiares de España (www.grupocistinosis.org) para favorecer su participación en el proyecto por medio de testimonios anónimos de pacientes y familiares.
Resultados: Elaboración de un documento-guía de transición coordinada, con propuestas concretas por especialidades y de mejora de la adherencia. El nefrólogo desempeña un papel clave en la transición en la cistinosis debido a la afectación renal que domina la patología, y al hecho de que la mayoría de los pacientes haya recibido un trasplante renal antes de la edad adulta. El documento se estructura en los siguientes apartados: Estrategia de la UE para enfermedades raras. Definición de la transisición y su importancia.
Particularidades de los adolescentes con cistinosis. Plan de transición: etapas y adquisición de competencias. Barreras para la transición en cistinosis. Agentes implicados y líderes de transición. El nefrólogo como coordinador de la transición. Gestor de casos. Propuesta de un modelo de transición y recomendaciones en nefrología, oftalmología, endocrinología, neurología y farmacia. Recomendaciones para mejorar la adherencia terapéutica en la transición.
Conclusión: se presenta un documento práctico que establece unas recomendaciones y un cronograma para guiar la transición de los adolescentes y adultos jóvenes con cistinosis en nuestro ámbito.
POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE: ¿A QUIÉN TRATAR?
M. FURLANO1, N. AYASREH1, L. ROCA ARGENTE2, A. RIUS PERIS3, T. MARTÍ4, E. ARS5, JA. BALLARÍN1, R. TORRA1
1NEFROLOGÍA. FUNDACIÒ PUIGVERT (BARCELONA), 2NEFROLOGÍA. HOSPITAL LA FE (VALENCIA), 3NEFROLOGÍA. HOSPITAL GENERAL UNIVERSITARIO DE CASTELLÓN (CASTELLÓN), 4RADIOLOGÍA. FUNDACIÒ PUIGVERT (BARCELONA), 5GENÉTICA Y BIOLOGÍA MOLECULAR. FUNDACIÒ PUIGVERT (BARCELONA)
Introducción: La poliquistosis renal autosómica dominante (PQRAD) es la enfermedad renal hereditaria más frecuente. El tolvaptán es el único tratamiento aprobado para la PQRAD. Aplicamos el algoritmo recomendado por el WGIKD de la EDTA 2016 para definir el porcentaje de progresadores rápidos (candidatos a tratamiento) de una consulta de enfermedades renales hereditarias (ERH).
Material y métodos: Se analizaron 206 pacientes consecutivos de una consulta de ERH de forma prospectiva según recomendaciones de la EDTA-WGIKD 2016. Se utilizó tanto ecografía con resonancia magnética nuclear (RMN) aplicado a la clasificación por imágenes para la PQRAD de la Clínica Mayo.
Resultados: El 39,80% se excluyeron para tratamiento por estar fuera de rango de edad y el 8.25 % por FG<45ml/m.
Clasificamos los 124 restantes en 3 grupos: Grupo 1 (18-30 años y FG>45ml/m) 19 pacientes, Grupo 2 (31-40 años y FG 45-90ml/m) 49 pacientes y el Grupo 3 (41-50 años y FG 45-60ml/m) 56 pacientes. Los del grupo 2 con FG>90ml/m y los del grupo 3 con FG>60ml/m se consideraron progresadores lentos. Mediante ecografía renal se valoró longitud renal >o< 16,5 cm: 17 de los 124 candidatos, se tratarían con tolvaptán por longitud >16,5 cm y menos de 45 años. En aquellos con longitud renal <16,5 cm y >16,5 cm entre 45 y 50 años, se analizó el volumen por ecografía y/o RMN según Clasificación Mayo. Por RMN 10 pacientes se tratarían. Mediante ecografía: 4 pacientes cumplen criterios 1D-1E y serían candidatos a tratamiento, pues aunque la ecografía sobreestime, la clasificación 1D-E no deja lugar a dudas. Los 17 pacientes del grupo 1A-1B tendrán conducta expectante. Los 1C por ecografía (9 pacientes) son probables candidatos pero debe hacerse RMN para aplicar correctamente la Clasificación Mayo. Por lo tanto el 17,47% del total de pacientes recibirán tratamiento a falta de reevaluar los clasificados como1C por ecografía (4.36%).
Conclusiones: La RMN es la prueba de imagen a utilizar para la fórmula Mayo. Es factible que la ecografía sea suficiente cuando se obtiene una clasificación 1A-B. Para 1C por eco recomendamos realizar RMN. En una consulta monográfica de ERH, el 52,42% de pacientes poliquísticos no se tratarán por estar fuera de rango de edad o FG<45ml/m. En base a los modelos de predicción trataríamos alrededor de un 20% de los pacientes.
PREVALENCIA DE FACTORES DE PROGRESION DEL DETERIORO DE LA FUNCION RENAL EN PACIENTES CON POLIQUISTOSIS RENAL AUTOSOMICA DOMINANTE
MT. JALDO RODRIGUEZ1, FJ. BORREGO-UTIEL1, E. MERINO GARCIA1, MJ. GARCIA CORTES1, M. POLAINA RUSILLO1, MM. BIECHY BALDAN1
1S. NEFROLOGÍA. COMPLEJO HOSPITALARIO DE JAEN (JAEN)
Objetivo: Analizar prevalencia de factores de riesgo para deterioro de función renal en pacientes con poliquistosis renal autosómica dominante (PQR) y su grado de relación con la función renal.
Metodología: Estudio transversal de pacientes con PQR seguidos en consulta. Primera analítica a partir 2007. GFR estimado con MDRD4. Tamaños renales: medición con ecografía coincidente con analítica.
Resultados: Incluimos 92 Pacientes, 44±15 años (15-78), 45 Varones (48,9%). Peso 73.4±15.2 kg (50-117), TAS 126±16 mm Hg (97-169) y TAD 78±11 mm Hg (52-106).
Función renal: Cr 1,3±0,6 mg/dl (mediana 1,2; rango 0,7-4,1), GFR 64±26 ml/min/1,73 m2, (mediana 63,3; rango 16,6-138 ml/min/1,73 m2). Estadíos ERC: 21,7% ERC1, 35,9% ERC2, 30,4% ERC3 y 12% ERC4. Proteinuria: 130±110 mg/gCr (mediana 94;rango 28-529). Prot/Cr>150 mg/gCr 17,8% y >500mg/gCr 2 pacs (2,2%). Albuminuria 34±56 mg/gCr (mediana 14;rango 1,56-348). Alb/Cr>30mg/gCr inicial 27,8% y >300 mg/gCr sólo 1 pac (1,1%).
Urico inicial 5,8±1,9 mg/dl (mediana 5,5;rango 2,3-12,7 mg/dl). Urico alto (>6 mg/dl): 38 pacs (41,3%). Con estadío ERC se incrementó edad (ERC1 31±12;ERC2 40±14;ERC3 52±10 y ERC4 60±11 años;p<0,001), ácido úrico (ERC1 5,0±1,7, ERC2 5,0±0,9, ERC3 6,5±2,2 y ERC4 7,8±1,3 mg/dl; p<0,001), proteinuria (ERC1 85±26, ERC2 100±78, ERC3 150±135 y ERC4 244±127 mg/Cr; p<0,001) y albuminuria (ERC1 15±3, ERC2 24±40, ERC3 48±81 y ERC4 61±50 mg/g Cr; p=0,032).
Tamaño renal (n=39 pacientes) fue 13,7±2,4 cm, mediana 13,4 cm;rango 10-18,5 cm;9 pacs con >16 cm (24,3%). Se correlacionó con Cr (r=0.459;p=0.003), úrico (r=0.485;p=0.002), GFR (r=-0.388;p=0.015). No asociación con número de hipotensores, con TAS o TAD.
Según niveles Cr (≤1.5, >1.5&≤2.5; >2.5 mg/dl) observamos: mayores niveles úrico (5.2±1.4; 7.3±2.4; 7.9±1.3 mg/dl;p<0.001), proteinuria/Cr (96±59; 211±169; 271±101 mg/gCr;p< 0.001), albuminuria/ Cr (20±31; 70±97; 77±43 mg/g Cr;p<0.001), mayor edad (40±14; 56±12; 59±10 años;p<0.001), menor GFR (75±21; 36±7; 18±1.3 ml/min/1,73 m2;p<0.001), mayor número de hipotensores (0.7±0.8; 1.1±0.6; 1.6±0.9,P<0.001).
Considerando niveles albuminuria (≤30 mg/gCr; >30&≤300 mg/gCr y >300 mg/gCr) observamos: diferencias en Cr (1,2±0,5; 1.7±0.8; 1.9 mg/dl;p=0.003), úrico (5,6±1,5; 6,1±2,2; 12,7 mg/dl;p=0.001) y GFR (68±25; 50±24; 42 ml/min1,73 m2;p=0.01).
Según niveles séricos de úrico (≤6, >6&≤8; >8 mg/dl) observamos: mayor Cr (1,1±0,3; 1,6±0,7; 2.0±0.7 mg/dl;p=0.001), mayor proteinuria (115±86; 128±118; 202±161 mg/gCr;p=0.046) y albuminuria (24±34; 32±42; 83±116 mg/gCr;p=0.004), mayor edad (40±15; 50±12; 52±14 años;p=0.002), menor GFR (75±23; 51±24; 44±25 ml/min/1,73 m2;p<0.001).
Medicación: alopurinol 9,8%; estatinas 23.9%; IECA 26,1%, ARA2 37%, calcioantagonistas 13%, otros 6,6%, tiazida 9,8% y furosemida 4,3%. Con IECA o ARA2 58 pacs (63.0%). Hipotensores: 35,9% sin hipotensores;50% tomaban 1; 9,8% tomaban 2 y 4,3% tomaban 3.
Considerando número hipotensores (0,1,2,3): presentaron mayor TAS (121±12; 126±17; 127±13; 160±10 mm Hg;p<0.001) y TAD (75±11; 79±10; 79±12; 93±11 mmHg;p=0.012), mayor Cr (1.0±0.2; 1.5±0.7; 1.4±0.7; 1.7±0.7 mg/dl;p=0.001), mayor úrico (4.8±1.3; 6.4±2.1; 6.0±1.3; 6.2±0.9 mg/ dl;p=0.002), mayor edad (35±12; 50±13; 19±17; 54±9 años;p<0.001), mayor proteinuria/Cr (p= 0.018). Pacientes con IECA o ARA2 presentaron mayores valores de Cr, úrico, proteinuria y TAS y menor GFR.
Conclusiones: En pacientes con PQR la función renal deteriorada se asocia con mayores niveles séricos de ácido úrico, mayor proteinuria y albuminuria, mayores cifras de presión arterial y necesidad de mayor número de antihipertensivos. Los llamados factores de progresión en PQR quizás sean más bien marcadores de lesión renal ya que acompañan a una función renal deterioarada y quizás no tengan significado pronóstico.
CORRELACION ENTRE EL VOLUMEN RENAL TOTAL Y EL FILTRADO GLOMERULAR EN LA POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE
RP. RAMON PECES1, MO. MARTA OÑATE2, EC. EMILIO CUESTA2, BR. BEGOÑA RIVAS1, CV. CRISTINA VEGA1, CP. CARLOS PECES1, RS. RAFAEL SELGAS1
1NEFROLOGÍA. HOSPITAL LA PAZ (MADRID), 2RADIOLOGIA. HOSPITAL LA PAZ (MADRID)
En la poliquistosis renal autosómica dominante (PQRAD) identificar la relación entre estructura y función resulta fundamental para monitorizar la evolución de la enfermedad y evaluar los efectos de cualquier tratamiento. El crecimiento de los quistes y del volumen renal total (VRT) se considera el mejor marcador para predecir el deterioro del filtrado glomerular (FG) y la progresión de la insuficiencia renal. En una cohorte de 182 pacientes con PQRAD se midió el VRT y htVRT (VRT ajustado por la altura). El FG se determinó por la creatinina sérica (Crs), MDRD y cistatina C (Cis C). Las características epidemiológicas de los pacientes fueron 81 varones (44,5%) y 101 mujeres (55,5%), la edad 40,3±12,7 (rango 14-79 años), el índice de masa corporal (IMC) 24,7±4,3 kg/m2, la altura170,8±9,5 cm, el VRT 1467±1035 ml, el htVRT 859±600 ml/m, la Crs 1,19±0,49 mg/dl, la Cis C 0,98±0,39 mg/l y MDRD 68,8±25,9 ml/min/1,73 m2. El VRT y htVRT se correlacionaron positivamente con Crs (r2 = 0,47, p<0,05) y Cis C (r2 = 0,68, p<0,05) y negativamente con MDRD (r2 = 0,46, p<0,05). Los varones tenían mayor VRT (1758±1265 vs.1233±730 ml, p<0,005) y mayor htVRT (995±727 vs.749±449 ml/m, p<0,05) que las mujeres. Los pacientes >40 años tenían mayor VRT (1818±1140 vs.1204±864 ml, p<0,001), mayor htVRT (1068±654 vs.702±505 ml/m, p<0,001) y peor MDRD (55±23 vs.78±23 ml/ min/1,73 m2, p<0,001) que los pacientes <40 años. Los pacientes con IMC>25 tenían mayor VRT (1843±1275 vs. 1203±723 ml, p<0,001), mayor htVRT (1068±728 vs. 712±437 ml/m, p<0,001) y peor MDRD (60±26 vs. 75±23 ml/min/1,73 m2, p<0,001) que los pacientes con IMC<25. Los pacientes con HTA tenían mayor VRT (1752±1063 vs. 758±513 ml, p<0,001), mayor htVRT (1028±614 vs. 463±283 ml/m, p<0,001) y peor MDRD (61±25 vs.87±16 ml/min/1,73 m2, p<0,001) que los pacientes sin HTA. Estos hallazgos confirman la existencia de una correlación entre estructura (VRT, htVRT) y función (MDRD) y sugieren que el mayor incremento de volumen de los quistes renales y por tanto de VRT y htVRT, y la mayor pérdida de función renal se asocian con el sexo masculino, mayor edad, mayor IMC y presencia de HTA.
ANÁLISIS CLÍNICO-EPIDEMIOLÓGICO DE PACIENTES AFECTOS DE POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE. EXPERIENCIA EN NUESTRO CENTRO
L. DE LA VARA INIESTA1, B. RINCÓN RUIZ1, C. HERNÁIZ VALENCIA1, MP. SIERRA BERMEJO1, G. MARTÍNEZ FERNÁNDEZ2, J. USÓN CARRASCO1
1NEFROLOGÍA. HOSPITAL VIRGEN DE LA LUZ (CUENCA), 2NEFROLOGÍA. HOSPITAL GENERAL DE ALBACETE (ALBACETE)
Introducción: La poliquistosis renal autosómica dominante es la enfermedad renal hereditaria más frecuente. Constituye entre un 6 y un 10% de la población en tratamiento renal sustitutivo (TRS), siendo una enfermedad con gran impacto social.
Objetivo: analizar y describir la experiencia en nuestro centro, evaluando la seguridad y eficacia de nuestra estrategia.
Material y método: Estudio observacional, descriptivo, retrospectivo. Incluimos todos los pacientes con poliquistosis renal seguidos en nuestro centro desde 1996 hasta la actualidad.
Resultados: Analizamos 101 pacientes de edad media 56,25±17,45 años, el 54,5% mujeres. El periodo medio de seguimiento fue de 14,54 años. 75,9% presentaban antecedentes familiares. Al diagnóstico, la edad y el FG medio fueron de 42,96 años y 73,45 ml/min respectivamente. Durante el seguimiento la hipertensión arterial fue la manifestación clínica más frecuente (71,92%). La edad media con FG<60 ml/min fue de 43,06 años. La prevalencia de hipercolesterolemia, hiperuricemia y diabetes mellitus fue de 50,9%, 24,6% y 14% respectivamente. Entre las manifestaciones extrarrenales más frecuentes, destaca la patología gastrointestinal presente en el 74% de los pacientes, predominando los quistes hepáticos (67,27%, más frecuente en mujeres, p<0,001), seguidos de la patología biliar (11%) y divertículos de colon (6,2%); Las infecciones urinarias (19,8%), más frecuentes en mujeres (p<0,001) y los CRU (12,3%). Los aneurismas cerebrales fueron infrecuentes (0,99%). La edad de inicio de tratamiento renal sustitutivo (TRS) fue de 64,15 años. Encontramos una considerable variabilidad fenotípica entre individuos de la misma familia y el inicio de TRS fue más precoz (p<0,001) cuando la enfermedad era transmitida por el padre. Se registró una media de 4,47 ingresos hospitalarios, con una estancia media de 4,66 días. 57 pacientes se encuentran en seguimiento ambulatorio, 14 en tratamiento dialítico, 21 trasplantados y 9 han fallecido. La esperanza de vida es de 60,71 años, siendo la principal causa de muerte la patología cardiovascular e infecciosa.
Conclusión: Nuestra experiencia es similar a la reportada en la literatura. La PQRAD es una enfermedad sistémica, con alta carga genética, cuyas manifestaciones clínicas más frecuentes son la HTA y la ERC. Destaca la patología gastrointestinal, las ITUs y los CRU como manifestaciones extrarrenales más frecuentes. La heterogeneidad fenotípica en las familias refleja mutaciones diferentes en el mismo gen o la influencia de otros factores genéticos o medioambientales, siendo el inicio de TRS más precoz si la transmisión es por vía paterna. La esperanza de vida es inferior a la población general.
¿HAY QUE REALIZAR MONITORIZACIÓN AMBULATORIA DE PRESIÓN ARTERIAL (MAPA) EN LA ELECCIÓN DE DONANTE DE VIVO PORTADOR DE SÍNDROME DE ALPORT AUTOSÓMICO RECESIVO?
D. MANZANO SÁNCHEZ1, V. MARTÍNEZ JIMÉNEZ1, IS. SAURA LUJÁN1, MJ. GONZÁLEZ SORIANO1, S. LLORENTE VIÑAS1, M. GIL MUÑOZ1, F. MORALES CARAVACA1, C. GARCÍA ARNEDO1, L. JIMENO GARCÍA1
1NEFROLOGÍA. HOSPITAL CLÍNICO UNIVERSITARIO VIRGEN DE LA ARRIXACA (MURCIA)
Introducción: Los pacientes con síndrome de Alport Autosómico Recesivo (SAAR) suelen precisar terapia renal sustitutiva (TRS) a los 25-30 años de edad siendo de elección el trasplante renal. La donación de vivo ofrece ventajas frente a la de cadáver.
Dado que el SAAR es una enfermedad hereditaria, nos planteamos si los pacientes portadores pueden ser donantes y cuáles serían los riesgos.
Presentamos la primera familia descrita en la literatura con 3 hijos afectos de SAAR que han recibido trasplante de donante vivo emparentado.
Material y Método: Estudio prospectivo de una familia con SAAR (3 donantes, 3 receptores) con seguimiento hasta 5 años para valorar la evolución tanto de los donantes como de los receptores y el riesgo de desarrollo de enfermedad renal postrasplante. Recogemos: edad, sexo, parentesco, función renal (antes y después del trasplante), sedimento urinario, factores de riesgo cardiovasculares (hipertensión arterial (HTA), diabetes mellitus, dislipemia, obesidad, fumador) y TRS. Se realiza pretrasplante audiometría, exploración oftalmológica; y MAPA pre y postrasplante.
Resultados: Caso 1: varón, 27 años, HTA, dislipemia, en hemodiálisis 1 año. Donante su madre de 48 años con creatinina (Cr) 0,71 mg/dl y sedimento urinario normal. A los 5 años de seguimiento receptor con Cr 1,16 mg/dl y donante de Cr 0,81 mg/dl sin complicaciones.
Caso 2: mujer, 29 años, HTA, dislipemia, en hemodiálisis. Donante su padre de 55 años, dislipemia, fumador, Cr 0,88 mg/dl y microalbuminuria. A los 3 años, la receptora presenta deterioro de la función renal con biopsia con proliferación mesangial, actualmente Cr 1,5 mg/dl. El donante mantiene Cr 1,24 mg/dl y microalbuminuria.
Caso 3: mujer, 33 años, HTA, dislipemia, Cr 4 mg/dl. Donante su tía materna (no portadora) de 49 años, fumadora con Cr 0,82 mg/dl y microalbuminuria. A los 3 años la receptora presenta Cr de 1,05 mg/dl y la donante de 1,1 mg/dl.
Se realiza MAPA de los donantes: en el caso 2 presenta HTA pre y postrasplante, aunque tensión arterial en domicilio es normal. Los otros dos con MAPA normal.
Conclusiones: La evolución de los pacientes ha sido favorable, destacando mayor deterioro de la función renal en el segundo caso tanto en donante como receptor que atribuimos a la HTA enmascarada del donante.
Recomendamos la realización de MAPA para descartar HTA que podría ser un factor de mal pronóstico. Los donantes heterocigotos del SAAR precisan una valoración completa pretrasplante aunque estén asintomáticos.
LOS FACTORES DE PROGRESION DEL DETERIORO RENAL EN PACIENTES CON POLIQUISTOSIS RENAL SON MAS BIEN MARCADORES QUE ACOMPAÑAN AL DETERIORO DEL FILTRADO GLOMERULAR
FJ. BORREGO-UTIEL1, MT. JALDO RODRIGUEZ1, E. MERINO GARCIA1, S. ORTEGA ANGUIANO1, C. MORIANA1, MC. SANCHEZ PERALES1
1S. NEFROLOGÍA. COMPLEJO HOSPITALARIO DE JAEN (JAEN)
Objetivo: Analizar qué factores influyen en ritmo de deterioro de función renal en pacientes con poliquistosis renal autosómica dominante (PQR).
Metodologia: Estudio retrospectivo pacientes con PQR seguidos en consulta de Nefrología de adultos. Corte basal primera analítica a partir de 2007 y última analítica en última revisión. GFR con MDRD4. Tamaños renales: medición con ecografía coincidente con la analítica basal.
Resultados: Incluimos 92 Pacientes, 44±15 años, 45 Varones (48,9%). Seguimiento 61±26 meses, mediana 66 meses (rango 12-107).
Durante seguimiento, observamos incremento de Cr (de 1,3±0,6 subió a 2,1±1,9 mg/dl;p<0,001), bajó GFR (de 65±30 a 54±32 ml/min/1,73 m2;p<0,001), ascendió proteinuria (de 128±111 a 347±614 mg/g Cr;p=0,001) y albuminuria (de 34±54 a 126±255 mg/gCr;p<0,001). Urico no cambió.
Estadíos ERC inicial : 21.7% ERC1, 35.9% ERC2, 30.4% ERC3 y 12% ERC4. Estadíos ERC final: 14.3% ERC1, 31.9% ERC2, 24.2% ERC3 y 12,1% ERC4 y 17.6% ERC5.
Creatinina subió más en estadíos ERC avanzados: ERC1 0,8±0,1 a 0,9±0,2 mg/dl (p=ns);ERC2 1,1±0,2 a 1,3±0,4 (p=0.015);ERC3 1.5±0.3 a 2.9±2.1 (p=0.001 );ERC4 2.6±0,7 a 4,9±1,9 (p<0.001).
Urico cambió irregularmente con estadíos: ERC1 5.0±11.7 a 5,2±1,4 (p=ns);ERC2 5,1±1,0 a 5,8±1,4 (p=0.008);ERC3 6.5±2.2 a 6.2±1.2 (p=ns);ERC4 7,8±1,3 a 6,5±1,2(p=0,06 ).
Proteinuria/Cr subió especialmente en estadíos avanzados: ERC1 85±26 a 102±59(p=ns);ERC2 98±80 a 118±95(p=0,05);ERC3 150±135 a 454±558(p<0.001); ERC4 241±134 a 1219±1219 (p<0.001).
Albuminuria/cr ascendió en estadíos avanzados: ERC1 15±15 a 22±26 (p=ns);ERC 24±42 a 35±64 (p=ns);ERC3 48±81 a 200±326 (p=0.002);ERC4 62±53 a 399±379 (p<0,001).
Proteinuria final guardó relación con estadío inicial (ERC1 103±56, ERC2 118±95, ERC3 454±558 y ERC4 1220±1220 mg/gCr;p<0,001) igual que albuminuria (ERC1 23±25, ERC2 35±64, ERC3 200±326 y ERC4 399±379 mg/gCr;p<0,001). Al final: Albuminuria<30 mg/gCr 56,2%, >30&<=300 mg/gCr 32,6% y >300 mg/gCr 11,2%. Proteinuria<=150 mg/gCr 58,4%, >150&<=500 mg/gCr 23,7% y >500 mg/gCr 17,9%.
Según terciles deterioro GFR (<-2.9; >-2.9 &<-1.02; >-1.02 ml/min/año) observamos niveles basales mayores de Cr (1,5±0,7; 1,5±0,7; 1,1±0,3 mg/dl;p=0,007), úrico (6,2±2,2; 6,3±1,8; 5,0±1,3 mg/dl;p=0,010) y discreta mayor albuminuria (51±75; 33±57; 19±18 mg/gCr;p=0,035) pero no proteinuria/Cr en pacientes con mayor ritmo de deterioro renal, sin diferencias en sexo,edad,peso,TAS o TAD.
Según número hipotensores (0,1,2,3): tuvieron peor función renal final (Cr 1.1±0.3; 2.7±2.2; 2.3±2.0; 3.0±2.1 mg/dl;p=0.001 ), mayor úrico (p=0.003); mayor proteinuria/Cr final (116±61; 460±773; 349±384; 899±880 mg/gCr;p=0.003); mayor albuminuria/Cr (27±30; 165±296; 131±179; 454±513 mg/g Cr;p=0.011).
Según tamaños renales observamos ascenso de Cr, proteinuria y albuminuria y descenso en GFR en pacientes con riñones ≥12,5 cm.
Con regresión lineal de variación inverso Cr encontramos como predictores: Cr y sexo femenino.
Conclusiones: En pacientes con PQR observamos mayor progresión de la insuficiencia renal en pacientes con peor función renal, mayor proteinuria, niveles más altos de ácido úrico y en sexo femenino. La proteinuria progresó más en pacientes con peor función renal. Los niveles de úrico no se modificaron al considerar el grado de insuficiencia renal inicial. Pacientes con riñones de mayor tamaño deterioran más la función renal. Muchos de los factores de progresión en PQR son marcadores de lesión renal, que están presentes cuando hay función renal deteriorada, no indicando precozmente quién deteriorará más la función renal.
HIPEROXALURIA PRIMARIA TIPO 1. MUTACIÓN EN EL GEN AGTX-I244T. EVOLUCIÓN A LARGO PLAZO
Y. PARODIS LOPEZ1, R. PAPOYAN2, E. INGLES TORRES3, MI. LUIS YANEZ4, E. SALIDO RUIZ5, N. SABLON GONZALEZ1, N. LORENZO VILLALBA1, V. GARCIA NIETO2
1NEFROLOGÍA. HOSPITAL UNIVERSITARIO DE GRAN CANARIA DR. NEGRIN (LAS PALMAS DE GRAN CANARIA), 2NEFROLOGÍA PEDIÁTRICA. HOSPITAL UNIVERSITARIO NTRA SRA CANDELARIA (SANTA CRUZ DE TENERIFE), 3NEFROLOGÍA. HOSPITAL DE SANTA LUCÍA (CARTAGENA, MURCIA), 4NEFROLOGÍA PEDIÁTRICA. HOSPITAL UNIVERSITARIO NTRA SRA CANDELARIA (SANTA CRUZ DE TENERIFE), 5ANATOMÍA PATOLÓGICA. COMPLEJO HOSPITALARIO UNIVERSITARIO DE CANARIAS (LA LAGUNA, TENERIFE)
Introducción: La hiperoxaluria primaria tipo 1 (HOP1) es una rara enfermedad de herencia autosómica recesiva, causada por mutaciones en el gen AGTX que codifica la enzima alanina glioxilato aminotransferasa, necesaria en la metabolización del glioxilato. Una actividad insuficiente de esa enzima conduce a un incremento en la conversión de glioxilato a oxalato. El riñón es el primer órgano afectado, dando lugar a la aparición de litiasis de repetición, nefrocalcinosis e insuficiencia renal precoz.
Pacientes y Métodos: Presentamos el primer estudio evolutivo a largo plazo de cuatro pacientes pediátricos diagnosticados de HOP tipo 1, portadores de la misma mutación. (Tablas 1 y 2).
Tabla 1 Variables clínicas y analíticas al diagnóstico de los cuatro pacientes con hiperoxaluria primaria
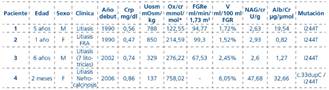
FRA: fracaso renal agudo. Ox/cr, NAG/cr y Alb/cr: cocientes urinarios calculados entre las concentraciones urinarias de oxalato, NAG y albúmina con respecto a la creatinina urinaria. V: volumen urinario corregido por 100 ml de FGR.
*Valores normales: 0-6 meses: 77-325 mmol/mol; 7-24 meses: 38-132 mmol/mol; 5-9 años: 22-70 mmol/mol
Tabla 2 Variables clínicas y analíticas en la última revisión de los cuatro pacientes con hiperoxaluria primaria

NS: ausencia de síntomas. *Valores normales: 9-12 años: 12-70 mmol/mol; adultos: 3-39 mmol/mol. **OMA:xx. En seguimiento por el servicio de Reumatología.
Resultados: Los 4 pacientes debutaron clínicamente en la primera década de la vida, tres con litiasis renal múltiple y uno con nefrocalcinosis, este último con deterioro de la función renal más severa en menos tiempo. Tras tratamiento con piridoxina, citrato potásico y sales de magnesio, han formado menos cálculos. Al diagnóstico, además de la hiperoxaluria, los cuatro pacientes tenían incremento del volumen urinario y tres de ellos defecto de la capacidad de concentración (Tabla 1).
Todos eran portadores en uno o ambos alelos de la mutación I244T que se asocia al polimorfismo L11P en el gen AGTX. Tres de los pacientes fueron homocigotos para la mutación, mientras que solamente una paciente (caso 4), la de peor evolución, mostró una mutación heterocigota compuesta (c.33dupC/ I244T). Hasta el momento no se han detectado signos de oxalosis sistémica.
Conclusiones: Los pacientes diagnosticados en la infancia que son portadores de la mutación “canaria” de HP1 muestran una evolución aceptable en respuesta al tratamiento farmacológico, a excepción de que debuten con nefrocalcinosis.
NEFROPATIA FAMILIAR ASOCIADA A UNA NUEVA MUTACIÓN DEL GEN DE LA UROMODULINA
C. ROSADO RUBIO1, MR. MANZANEDO RUBIO1, J. MARTIN-CENTELLAS1, E. BUENO MARTINEZ2, C. FELIPE FERNANDEZ1, MB. ALAGUERO DEL POZO1, J. MARTIN-GARCIA1, R. GONZALEZ SARMIENTO2
1NEFROLOGÍA. HOSPITAL NTRA SRA DE SONSOLES (AVILA), 2IBSAL Y LABORATORIO DE MEDICINA MOLECULAR. UNIVERSIDAD DE SALAMANCA (SALAMANCA)
La Enfermedad renal asociada a alteraciones de la UROMODULINA (Uromodulina Kidney Disease -UKD- en terminología KDIGO) es un subtipo de Enfermedad Renal Tubulointersticial Autosómica Dominante. Es la consecuencia de mutaciones en el gen UMOD (16p12), que codifica la proteína UROMODULINA (glicoproteína de Tamm-Horsfall). La expresión clínica más frecuente incluye hiperuricemia y Enfermedad Renal Crónica progresiva, sin proteinuria ni hematuria, riñones ecográficamente normales o con quistes medulares (denominada previamente Enfermedad Medular Quística) y fibrosis tubulointersticial inespecífica. Las mutaciones patogénicas se concentran en los exones 3, 4, 5 y 7.
Pacientes y métodos: Estudiamos una familia con diagnóstico clínico de UKD. Realizamos una búsqueda de mutaciones en el gen UMOD en el caso índice mediante hibridación molecular del DNA, previa amplificación del mismo mediante reacción en cadena de la polimerasa.
El caso índice es un varón de 49 años con creatinina de 2,4 mg/dl, con leve hiperuricemia (7,8-9,5 mg/dl) 4 años antes. El sedimento urinario, la microalbuminuria y el protocolo glomerular fueron normales. La biopsia renal mostró 9 glomérulos, 2-3 esclerosados y fibrosis periglomerular el resto, sin lesiones glomerulares, inflamación intersticial multifocal y fibrosis intersticial con atrofia tubular moderadas; inmunofluorescencia negativa. En los familiares constan: padre fallecido en programa de HD con trasplante previo, cuatro tías paternas fallecidas en programa de Hemodiálisis, abuelo paterno fallecido con ERC, y bisabuelo fallecido joven.
El estudio genético detectó la variante c.202G>C en el exón 3, en heterocigosis, que da lugar al cambio p.Glu68Gln (p.E68Q) (glutamina por glutámico en la posición 68 de la uromodulina). El análisis in silico de la variante la señala como probablemente patogénica: PolyPhen2, probablemente dañina (HumVar; score: 0,998); Mutation Taster Disease causing (probabilidad: 0,9971); MutPred probability of deletereus Mutation: 0,855, por lo que la catalogamos como mutación missense.
El estudio genético a familiares vivos reveló: 3 hermanos, 2 mujeres y 1 varón no afectados; y 1 hermano portador de la variante junto con sus 2 hijos, sin enfermedad renal actual. Dos primos del caso índice, de 55 a. con ERCA en prediálisis y otro de 52 a. portador de trasplante anticipado presentan la variante genética.
Conclusiones: Caracterizamos la mutación c.202G>C en heterocigosis del exón 3 del gen UMOD como la mutación missense patogénica causante de la UKD que padece la familia estudiada. En enfermedades renales no filiadas y un patrón hereditario es conveniente realizar estudios genéticos para determinar una posible nefropatía hereditaria y prevenir la progresión y afectación de individuos portadores.
FACTORES DE PROGRESIÓN DE INSUFICIENCIA RENAL EN UNA POBLACIÓN DE PACIENTES CON POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE (PQRAD)
L. ROCA ARGENTE1, Y. MANZUR AGUILAR1, T. BENKIRAN1, L. MUÑOZ SALVADOR1, JL. MOLL GUILLEM1, A. GALLARDO PÉREZ1, J. BROSETA MONZÓ1, J. HERNANDEZ JARAS1
1NEFROLOGÍA. HOSPITAL LA FE (VALENCIA)
Introducción: La PQRAD es la enfermedad renal hereditaria más frecuente con una prevalencia estimada de 1 de cada 800 pacientes y representa entre el 6-10% de la población en diálisis o con injerto renal funcionante. Se caracteriza por la progresión de quistes renales y aumento del tamaño renal que condiciona la aparición de enfermedad renal crónica generalmente en la edad adulta. La progresión depende de factores genéticos (PKD1, PKD2) y, en menor grado, de factores ambientales.
Objetivo: Estudiar factores de progresión de la insuficiencia renal con PQRAD.
Material y métodos: Estudio descriptivo retrospectivo de una población de 80 pacientes en seguimiento desde 2004 a 2014 . Se recogieron datos basales demográficos (edad, sexo, IMC), factores ya conocidos de riesgo cardiovascular (HTA, DM, dislipemia, tabaquismo, hiperuricemia, FGe < 60 ml/min/m2) y datos de progresión renal mediante aparición de evento definido por entrada en terapia renal sustitutiva (hemodiálisis, diálisis peritoneal o trasplante renal) o descenso del 50% o más del filtrado glomerular estimado según fórmula CKD-EPI.
Resultados: El promedio de edad era de 33,99 ± 13,3 con una mediana de seguimiento de 96 meses (mín 12-máx 120). De los pacientes que desarrollaron evento renal (21) el 90.5% eran hipertensos (p=0,006), el 52,4% eran dislipémicos (p=0,024) y el 47,6% eran fumadores (p=0,26). El descenso medio de FG anual estimado por fórmula CKD-EPI fue de 2,31 ml/min/ m2. Los pacientes que llegaron al evento presentaban una disminución anual de 4,84±2.5 ml/min/m2, mientras que en aquellos que no llegaron al evento fue de 1.4±2.94 ml/min/m2 (p<0.001). Al inicio de seguimiento, los pacientes que presentaron posteriormente evento renal presentaban mayores cifras de ácido úrico (6.32±1.59 vs. 4.72±1.55 mg/dl. p <0,05), un menor filtrado glomerular (42.06±22.29 vs. 97±26.28 ml/min. P<0.001) y mayor proteinuria (0.25 vs. 0.15 gr/día. p<0,05). En el análisis multivariante el FGe basal, la dislipemia y la proteinuria fueron variables independientes de mal pronóstico renal (p<0,001, p=0,014 y p=0,019 respectivamente).
Conclusiones: La PQRAD es una enfermedad hereditaria quística renal cuya progresión a la insuficiencia renal puede verse modificada por factores independientes de riesgo cardiovascular. A favor de lo publicado en la literatura, en nuestra población observamos que la proteinuria basal, dislipemia y filtrado glomerular son factores de progresión de daño renal.
GAMMAGRAFÍA CON GALIO-67: DIAGNÓSTICO DE INFECCIÓN DE QUISTE EN POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE (PQRAD)
E. CASILLAS SAGRADO1, M. DELGADO YAGÜE1, LV. BLANCO ANDREWS1, E. YEROVI LEÓN1, M. RIVERA GORRÍN1, V. BURGUERA VION1, F. LIAÑO1
1NEFROLOGÍA. HOSPITAL RAMÓN Y CAJAL (MADRID)
Introducción: En las recientemente publicadas guias clínicas de la PQRAD se establece como diagnóstico defínitivo de infección de quiste renal la obtención de material intraquístico compatible con infección. La prueba de imagen de elección que se propone es el PET-TAC, que es una prueba costosa y poco disponible. La Gammagrafía con leucocitos marcados con Tc-99, por otro lado, requiere la extracción previa de hemoderivados y es engorrosa. En este trabajo queremos resaltar la utilidad de la GA-67c en el diagnóstico de la infección de quiste en una serie de casos ocurridos en el último año.
Material y métodos: Entre enero de 2015 y enero de 2016, tres mujeres y dos hombres con PQRAD, mediana edad 43 años (rango 33-72 años), consultaron por fiebre (mediana de 39ºC), cuatro referian dolor.
Resultados: La ecografía renal detectó infección en 1 de los 5 casos y el TC en 2 de 5. La GA-67c fue positiva en 4 casos y negativa verdadera en 1. La GA-67c se usó además para decidir la duración en el seguimiento de los pacientes. La GA-67c fue diagnóstica en 5/5 casos (Valor diagnótico del 100%) con rendimiento muy superior al TC (2/5) y la ecografía (1/5). Además permitió realizar el diagnóstico definitivo en aquellos pacientes en los que el resultado de la ecografía y el TC eran opuestos.
Conclusiones: La GA-67c es una prueba con gran disponibilidad, no invasiva, disponible en la mayoria de los centros y que puede realizarse en todos los casos de PQRAD con sospecha de infección de quiste y tiene un coste bajo. Por todo esto consideramos que debería incluirse dentro de las pruebas de elección en el diagnóstico de la infección de quiste en la PQRAD.

ENFERMEDAD DE FABRY: COMPROMISO RENAL AL MOMENTO DEL DIAGNÓSTICO EN UNA POBLACIÓN DE 60 PACIENTES EN ARGENTINA
FP. FERNANDO PERRETTA1, SJ. SEBASTIAN JAURRETCHE2, NA. NORBERTO ANTONGIOVANNI3
1TERAPIA INTENSIVA. HOSPITAL ENRIQUE ERILL (ESCOBAR, PROVINCIA DE BUENOS AIRES, ARGENTINA), 2NEFROLOGÍA. INSTITUTO UNIVERSITARIO ITALIANO (ROSARIO, PROVINCIA DE SANTA FE, ARGENTINA), 3NEFROLOGÍA. INSTITUTO DE NEFROLOGÍA PERGAMINO (PROVINCIA DE BUENOS AIRES, ARGENTINA)
La comunicación corresponde a un grupo de trabajo o un estudio multicentrico:
Miembros de GINEF Argentina (Grupo de Investigación Nefrológica en la Enfermedad de Fabry)
Introducción: la enfermedad de Fabry (EF) es un trastorno poco frecuente, hereditario, ligado al cromosoma X, ocasionado por el déficit de la enzima alfa-galactosidasa A (a-gal-A). Esto determina el depósito de glucoesfingolipidos neutros (principalmente globotriaosilceramida - Gb3) en los lisosomas de diferentes tipos celulares. Es una enfermedad multisistémica que afecta a ambos sexos, y que ocasiona una serie de complicaciones graves predominantemente a nivel neurológico, renal y cardiovascular, que reducen la expectativa y la calidad de vida. Específicamente el compromiso renal se manifiesta con albuminuria, proteinuria e insuficiencia renal progresiva, hasta llegar a requerir de tratamiento sustitutivo renal.
Material y método: se analizaron 60 pacientes con diagnóstico de EF en Argentina. La actividad de la α-galactosidasa-A se realizó en papel de filtro por método fluorométrico. El estudio mutacional mediante MPLA y secuenciación. Se calculó el filtrado glomerular (FG) por fórmulas MDRD y CKD/EPI en los pacientes adultos, y Schwartz en pediátricos. Se consideró albuminuria a valores > a 20 ug/min en orina minutada o > a 30 mg/g de creatinina en orina al azar, y proteinuria a valores > a 150 mg/d, todo lo previo en al menos dos muestras diferentes.
Resultados: se evaluaron 60 pacientes de 9 genotipos diferentes; 35 mujeres y 25 varones, con una edad promedio de 26.6 y 22.8 años respectivamente. Se identificaron 10 casos índices; 8 masculinos y 2 femeninos, todos adultos, 6 de ellos con algún grado de fallo renal, 7 con albuminuria y/o proteinuria, y 4 en plan de diálisis. De los 37 pacientes adultos evaluados: 6 varones (16.2%) presentaron IRCt al momento del diagnóstico, 6 hombres y 10 mujer presentaron FG < a 90 ml/min (43.2%), y 5 mujeres y 3 varones (21.6%) valores de hiperfiltración glomerular.
Albuminuria fue detectada en 12 mujeres y 1 hombres (35.1%), y 10 varones y 2 mujeres presentaron proteinuria (32.4%). En los 23 pacientes pediátricos se destaca hiperfiltración en 11 de ellos (47.8%), y 3 pacientes presentaron albuminuria (13%).
Conclusiones: el compromiso renal en la EF se evidencia desde edades tempranas con la compensación glomerular (hiperfiltración), la que puede enmascarar el deterioro orgánico. La albuminuria si bien se observa en niños, es de mayor prevalencia en edades adultas, donde un tercio de los pacientes presentaron proteinuria. Los resultados muestran un alto compromiso renal en la población adulta, siendo más temprano y severo en varones con EF.
SIGNOS Y SÍNTOMAS PARA EL RECONOCIMIENTO PRECOZ DE LA ENFERMEDAD DE FABRY, DIFERENCIAS DEL CRIBADO SEGÚN ESPECIALIDADES MEDICAS
L. VELÁZQUEZ RÍOS1, A. PUENTE GARCÍA2, H. MAGRO GARCÍA1, L. LOZANO MANEIRO2, L. ALEGRE ZAHONERO2, J. RUIZ RUIZ1, A. ZAPATERO GAVIRIA1
1MEDICINA INTERNA. HOSPITAL UNIVERSITARIO DE FUENLABRADA (MADRID), 2NEFROLOGÍA. HOSPITAL UNIVERSITARIO DE FUENLABRADA (MADRID)
Introducción: La enfermedad de Fabry es un trastorno innato metabólico secundario a la ausencia/deficiente actividad de la a-D-galactósido galactohidrolasa, como resultado se produce la acumulación progresiva de globotriaosilceramida (Gb3/GL-3).
Material y método: Se realizó un análisis descriptivo basado en la revisión de historias clínicas y criterios que llevaron a los especialistas a solicitar el cribado de Enfermedad de Fabry desde 2005 hasta 2016. Se recogieron datos demográficos, clínicos y analíticos. También se valoró la realización del estudio genético confirmatorio. El análisis estadístico se realizó con el software IBM.SPSS-V-22.
Resultados: Se han solicitado 42 determinaciones de actividad de αgalactosidasa. El 45,2% eran mujeres y el 54,8% hombres, siendo la media de edad de 54,5 años.
Se objetivó mutación genética en el 12% de la muestra (n=5), de los cuales 3 eran mujeres. Actualmente la terapia de reemplazo enzimática la recibe solo un paciente.
La distribución por frecuencia de la solicitud del despistaje destaca Nefrología siendo responsable del 81% de las peticiones seguido de Medicina Interna con un 10%, de forma minoritaria Neurología, Pediatría, Cardiología y Reumatología.
En cuanto a la función renal, el 52,4% presentaban proteinuria, de los cuales el 47,7% en rango no nefrótico. El filtrado glomerular, en los tres meses previos al cribado fue el 23,8% FG>90ml/min, el 21,4% 60-89ml/min, el 28,6% 30-59ml/min y un 16,7% con FG de 15-29ml/ min ml/min/1.73m2. Tan solo el 2,4 % presenta un FG<15ml/min.
La manifestación neurológica más frecuente fue las parestesias presentes en el 16,7% de los pacientes. Un 4,8% (n=2) de la muestra presentó ictus. Se realizo cribado dos pacientes por angioqueratoma corporis difuso.
Se realizó ecocardiograma al 57,2% de los pacientes, de los cuales el 16,7% presentaban hipertrofia del ventrículo izquierdo. De la muestra, el 9,5% presentó insuficiencia cardíaca. Valvulopatía14,7% y 9,5% presentan arritmias. La valoración oftalmológica se realizó en el 66,6% de los pacientes, presentando cornea verticillata el 9,5%.
Discusión y conclusiones: Destaca que casi la mitad de los pacientes no tienen ecocardiograma, como prueba complementaria para el diagnóstico de la hipertrofia ventricular, teniendo en cuenta que se trata de una manifestación frecuente con implicaciones pronósticas, se debería fomentar entre las pruebas de sospecha diagnostica.
La enfermedad de Fabry, continúa siendo infradiagnosticada. En los últimos años ha aumentado el número de pruebas de cribado, especialmente en adultos, siendo nefrología el máximo peticionario en nuestro centro, que consideramos pueda ser por la presencia de proteinuria como parámetro objetivo de la enfermedad. Educar en el cribado de casos de sospecha clínica, especialmente en adultos jovenes en otras especialidades médicas es un punto a mejorar.
ENFERMEDAD DE FABRY CLÁSICA EN PEDIATRÍA: COMPROMISO RENAL AL MOMENTO DEL DIAGNÓSTICO EN UNA POBLACIÓN DE 21 NIÑOS EN ARGENTINA
NR. ANTONGIOVANNI1, S. JAURRETCHE2, F. PERRETTA3
1NEFROLOGÍA. INSTITUTO DE NEFROLOGÍA PERGAMINO (PERGAMINO), 2NEFROLOGÍA. INSTITUTO UNIVERSITARIO ITALIANO DE ROSARIO (ROSARIO), 3NEFROLOGÍA. HOSPITAL ENRIQUE ERILL DE ESCOBAR (ESCOBAR)
La comunicación corresponde a un grupo de trabajo o un estudio multicentrico:
Miembros de GINEF Argentina (Grupo de Investigación Nefrológica en la Enfermedad de Fabry).
Introducción: La Enfermedad de Fabry (EF) es una patología hereditaria, ligada al cromosoma X, cuyo defecto genético ocasiona el déficit total o parcial de la enzima Alfa-Galactosidasa A (Agal A) con el consecuente depósito de glicoesfingolípidos neutros en los lisosomas celulares. El compromiso de riñón, cerebro y corazón en su evolución, acorta la expectativa de vida por disfunción multiorgánica. En EF se han descripto más de 900 mutaciones, que se clasifican en clásicas (de compromiso generalizado y temprano) o tardías. Aun así, en la actualidad, no está claro a qué edad comienza el compromiso severo e irreversible de estos órganos diana.
Material y método: Se analizaron 21 pacientes entre 2 y 16 años (media: 7 años) con EF y Mutación Clásica Severa (E398x y L415p). Todos diagnosticados por screening familiar y corroborados por estudio mutacional mediante MPLA y secuenciación.
Se consideraron signos y síntomas relacionados a EF al momento del diagnóstico. Para evaluar compromiso renal se calculó el filtrado glomerular (FG) por fórmula de Schwartz, interpretándose hiperfiltrado cifras > 140 ml/min; se consideró albuminuria a valores > a 18 ug/min en orina minutada o > a 30 mg/g de creatinina en orina al azar.
Resultados: Del total de los pacientes pediátricos (12 mujeres y 9 varones), el 50% de los mismos (11/21) presentó hiperfiltración (FG>140 ml/min), como primer paso en la progresión a albuminuria y enfermedad renal progresiva. Solo 2 presentaron albuminuria pero no proteinuria. De estos 11 pacientes con compromiso renal incipiente (9 mujeres entre 5 y 16 años, y 2 varones entre 10 y 16 años); 9 presentaban otros signos relacionados a EF: angioqueratomas (5 pacientes), acroparestesias (5 pacientes), bradicardia sinusal (2 pacientes), y una paciente presentaba compromiso de SNC.
Apenas 3 mujeres menores a 7 años NO presentaron sintomatología relacionada a la EF.
Conclusión: La EF en su variante clásica es una enfermedad progresiva, con menor expectativa de vida en edad adulta por compromiso de órganos vitales.
La hiperfiltración debe considerarse un signo incipiente de nefropatía.
En familias con mutaciones severas (con nula, o casi nula actividad enzimática) debemos considerar todo posible marcador de daño en órganos vitales desde edad pediátrica, dado que el diagnóstico preciso permitirá un tratamiento temprano y eficaz.
ENFERMEDAD DE FABRY. TERAPIA DE SUSTITUCIÓN ENZIMÁTICA EN PACIENTES EN HEMODIÁLISIS
S. JAURRETCHE1, F. PERRETTA1, N. ANTONGIOVANNI1
1DOCTORADO EN CIENCIAS BIOMÉDICAS. INSTITUTO UNIVERSITARIO ITALIANO DE ROSARIO (ROSARIO)
Introducción: La enfermedad de Fabry (EF) es una enfermedad de depósito lisosomal, de herencia ligada al cromosoma X, resultante del déficit de actividad enzimática α-galactosidasa-A (α-gal-A). El depósito de glicoesfingolípidos en el parénquima renal produce nefropatía proteinúrica progresiva. La enfermedad renal crónica terminal (ERCT) ocurre generalmente en la cuarta década de la vida en varones afectados y es una de las principales causas de mobimortalidad en ambos sexos. En pacientes con EF en diálisis la principal causa de mortalidad son los eventos cardio y cerebrovasculares, resultando en una menor supervivencia comparados con pacientes no diabéticos. El propósito del presente trabajo es evaluar la eficacia de la terapia de reemplazo enzimático (TRE) sobre la incidencia de dichos eventos, que aumentan la mortalidad de los pacientes con EF en diálisis.
Material y métodos: De nuestra población de pacientes con EF se incluyeron los que recibieron diálisis durante su evolución. Se realizó un análisis retrospectivo de la historia clínica de los mismos. Se consideró evento cardiovascular a: arritmia en ECG, cambios ecocardiográficos típicos o ataque cardíaco clínicamente evidente. Evento cerebrovascular se definió como cambios típicos en RMN de SNC o Stroke clínicamente evidente. Los datos se analizaron en base estadística SPSS.
Resultados: Se estudiaron 7 varones de 6 genotipos (mutaciones clásicas del gen GLA). Datos epidemiológicos se resumen en la Tabla 1. En 5 pacientes que recibieron TRE durante diálisis no hubo efectos adversos. Los 2 pacientes que no recibieron TRE fallecieron. Un paciente falleció de causa cardiovascular recibiendo dosis de 0,2 mg/Kg. Un evento cardiovascular no mortal fue registrado en un paciente recibiendo dosis de 1 mg/Kg.
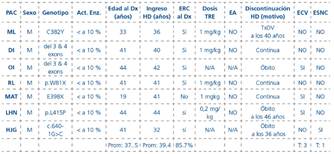
Conclusiones: Aun cuando nuestros resultados no permiten conclusiones definitivas, podríamos concluir que la TRE es segura en pacientes en diálisis y eficaz en prevenir eventos cardio y cerebrovasculares dependiendo de la dosis administrada.
DEVELOPMENTAL SWITCH FOR POLYCYSTIC HEPATIC AND KIDNEY DISEASE (PKD) TREATMENT: UNMASKING THE SIGNALING PATHWAYS THAT TEMPORALLY AND SPATIALLY DRIVE CYST INITIATION AND CYST PROGRESSION
O. LAMAS GONZALEZ1, S. BRAVO2, A. CORDIDO EIJO1, AB. SANZ3, A. ORTIZ3, T. WATNICK4, GG. GERMINO5, C. DIAZ6, MA. GARCIA GONZALEZ1
1GRUPO DE GENÉTICA Y BIOLOGÍA DEL DESARROLLO DE LAS ENFERMEDADES RENALES. INSTITUTO DE INVESTIGACIÓN SANITARIA DE SANTIAGO (IDIS) (SANTIAGO DE COMPOSTELA), 2UNIDAD DE PROTEÓMICA. INSTITUTO DE INVESTIGACIÓN SANITARIA DE SANTIAGO (IDIS) (SANTIAGO DE COMPOSTELA), 3LABORATORIO DE NEFROLOGÍA. IIS-FUNDACIÓN JIMENEZ DÍAZ (MADRID), 4NEPHROLOGY. UNIVERSITY OF MARYLAND SCHOOL OF MEDICINE (BALTIMORE, MA, US), 5NATIONAL INSTITUTE OF DIABETES AND DIGESTIVE AND KIDNEY DISEASE. NATIONAL INSTITUTE OF HEALTH (BETHESDA, MA, US), 6NEFROLOGÍA. HOSPITAL CLINICO DE SANTIAGO DE COMPOSTELA (SANTIAGO DE COMPOSTELA)
The pathogenesis of Polycystic Kidney Disease (PKD) has been related to a number of different mechanisms that make it very complex and there is no therapy for complete inhibition of cystogenesis, although there are advances in controlling cyst volume and cyst progression.
Taking advantage of the identified developmental window in PKD using the Pkd1 conditional KO mouse, we have previously shown the identification of the differential proteome of the cystic and non-cystic Pkd1 mutant kidneys, as well as those pathways associated to the cystic progression under inflammatory respond.
Here, we further explore the effect of therapeutically targeting our very short list of 16 candidates in ADPKD prevention and control. During cystogenesis, we described sequentially altered cell morphogenesis, ECM-cell interactions and cell metabolism. In this regard, we have targeted the identified pathways both inhibiting complete cystogenesis deriving from distal nephron segment, as well as, reducing/delaying global cystic progression and recovering kidney and liver function. Indeed, drug targeting of our candidates, comparison and/or combination of targeting with those partially approved, leaded to surprisingly results not only in cyst control but also in unexpected phenotypical effects.
In conclusion, we have first described the proteome related to ADPKD, puzzling out the main 3 hits that likely take place throughout the outcome of the disease, from PKD deficiency to kidney injury and inflammation-dependent tissue remodeling processes, and revealing different signaling pathways that could be temporally and spatially driven cyst initiation and progression.
Moreover, we also showed that an effective treatment, that controls both origin and progression of ADPKD, relies on drug combination of therapeutics targeting different pathways and cystic origins.
TWEAK UNMASKS A NEW MOLECULAR MECHANISM OF DISEASE PROGRESION IN AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
O. LAMAS GONZALEZ1, AB. SANZ2, I. ROWE3, A. CORDIDO EIJO1, A. BOLETTA3, A. ORTIZ2, MA. GARCIA GONZALEZ1
1GRUPO DE GENÉTICA Y BIOLOGÍA DEL DESARROLLO DE LAS ENFERMEDADES RENALES. INSTITUTO DE INVESTIGACIÓN SANITARIA DE SANTIAGO (IDIS) (SANTIAGO DE COMPOSTELA), 2LABORATORIO DE NEFROLOGÍA. IIS-FUNDACIÓN JIMENEZ DÍAZ (MADRID), 3LABORATORY OF MOLECULAR BASIS OF POLYCYSTIC KIDNEY DISEASE. SAN RAFFAELE SCIENTIFIC INSTITUTE (MILAN, ITALIA)
The pathogenesis of Polycystic Kidney Disease (PKD) appears to involve altered tubular cell proliferation, cell death, differentiation and polarity as well as inflammatory and pro-fibrotic factors. It has been reported that acute kidney injury (AKI) and inflammation accelerate cystogenesis and TWEAK, a TNF-like cytokine, has a key role in AKI and renal inflammation.
We have previously shown through a mouse model for conditional inactivation of PKD1 gene that timing is vital to modulate response to treatment. In our model, TWEAK appeared as a window dependent modulator of cystogenesis, inhibiting cystic progression in the cystic window, and/or promoting cystogenesis in the non-cystic window.
Here, we studied the TWEAK-dependent mechanisms that are likely underlying these differential phenotypes and also test the effect of this cytokine in a mouse model of very aggressive ADPKD early-onset. Analysis of the main cell signaling pathways revealed that TWEAK could be promoting differential proliferation through ERK/mTOR/MAPK cascade, whilst apoptosis seemed not to be deeply involved. Inflammation background appeared to play also an important role under TWEAK administration in these conditions. Indeed, our preliminary results are pointing out that ratio between macrophage subsets could be swinging tissue remodeling and immune response to kidney injure, and thus, modulating the development and progression of ADPKD.
Our results further reinforce the developmental impact of PKD1 inactivation and TWEAK in cystogenesis, and the importance of detection of the right time to treatment. Our data are also highlighting the contribution of inflammation and immune system to ADPKD, leading to new regulatory mechanisms that modulate the outcome of the disease and could be targets in disease control.
PAPEL DE LA HIPERTENSION ARTERIAL EN EL SINDROME HEMOLITICO UREMICO ATIPICO
M. MACIA1, A. JARQUE1, V. CASTRO2, C. GARCIA-CANTON3, N. DEL CASTILLO1, F. VALGA4, T. MONZON4, MD. GETINO1, JM. GONZALEZ-POSADA1
1NEFROLOGÍA. HUNS DE LA CANDELARIA (SANTA CRUZ DE TENERIFE), 2ANATOMIA PATOLOGICA. HUNS DE LA CANDELARIA (SANTA CRUZ DE TENERIFE), 3NEFROLOGÍA. CHU MATERNO INSULAR (LAS PALMAS DE GRAN CANARIA), 4NEFROLOGÍA. CLINICA QUIRON (SANTA CRUZ DE TENERIFE)
El SHUa se caracteriza por la triada anemia hemolítica microangiopática no inmune (MAT), trombocitopenia y fracaso renal agudo. El tratamiento con eculizumab ha mejorado el pronóstico de estos pacientes, su administración precoz es uno de los factores que en mayor medida ha contribuido a este efecto, es necesario un alto grado de sospecha para agilizar el diagnóstico. Se ha descrito la presencia de HTA tanto en el debut como durante su evolución y en ocasiones no se acompaña de alteraciones hematológicas, lo que puede retrasar el diagnóstico. Presentamos nuestra experiencia con 5 pacientes con SHUa, analizamos la HTA y su manejo como hallazgo común.
Entre Mayo 2013 y Enero 2016 se diagnosticaron 5 pacientes de SHUa. 4 H/1 M; edad 22-60 años. Todos debutaron con MAT, actividad ADAMTS-13 normal y 3 presentaron un evento trigger. 4 tenían función renal previa normal, precisaron plasmaféresis (entre 1-10 sesiones) y todos diálisis. La biopsia renal confirmo el diagnostico (n=4; 1 tenía lesiones de cronicidad) y todos tenían lesiones vasculares de HTA. Estudio genético (n=4): haplotipo de riesgo MCP (n=3) y delección CFHR2 (n=1). 4 recibieron eculizumab (tiempo medio para inicio 18 d; 1-50 d), la respuesta hematológica fue completa y 3 recuperaron función renal (el tiempo hasta no precisar diálisis (10-75 d). de los 4 pacientes en 2 se suspendió el tratamiento (por respuesta hematológica y renal completa en uno y daño renal crónico en otro). En el diagnóstico todos presentaron HTA grado III de novo (media 190/100) y precisaron 3-6 fármacos para su control. Retinopatía hipertensiva: n=3 (grados I-III); HVI por ecocardiograma: n=3 (severa en 2). Tras un periodo variable (1-3 meses) los que recibieron eculizumab la HTA se controló (1 no precisa tratamiento). El paciente que no se trató persiste sin control y 4 fármacos.
El eculizumab permitió el control mantenido de la MAT en todos los pacientes. La recuperación de la función renal se produjo en aquellos con función renal previa normal, inicio más precoz del tratamiento y sin lesiones de cronicidad en la biopsia. La HTA grado III fue un hallazgo común en el debut del SHUa. Todos presentaban diferente grado de afectación de órganos diana y dificultad para alcanzar cifras de TA adecuadas. La respuesta al eculizumab parece acompañarse de un mejor control de la HTA. En aquellos pacientes que desarrollen HTA severa se deberían realizar estudios para detectar la presencia de un posible SHUa.
HIPERTENSIÓN ARTERIAL SECUNDARIA A SÍNDROME DE GORDON: PRESENTACIÓN DE UNA GRAN FAMILIA ESPAÑOLA CON UNA MUTACIÓN DEL GEN WNK1
RP. RAMON PECES1, RM. ROCIO MENA2, BR. BEGOÑA RIVAS1, CV. CRISTINA VEGA1, CP. CARLOS PECES1, RS. RAFAEL SELGAS1, JN. JULIAN NEVADO2
1NEFROLOGÍA. HOSPITAL LA PAZ (MADRID), 2GENETICA. HOSPITAL LA PAZ (MADRID)
El síndrome de Gordon (o pseudohipoaldosteronismo tipo 2, o hipertensión hiperpotasémica familiar) es una causa rara de hipertensión arterial secundaria con herencia autosómica dominante que se manifiesta en la infancia. Se caracteriza por hipertensión, acidosis metabólica hiperpotasémica e hiperclorémica, baja renina y niveles habitualmente normales de aldosterona, que es sensible a diuréticos tiazídicos. La enfermedad se produce por mutaciones en al menos 4 genes: WNK1, WNK4, KLHL3 y CUL3. Existe una correlación genotipo-fenotipo, con las mutaciones de CUL3 con un fenotipo más severo y con las mutaciones de WNK1 con un fenotipomenos severo, en términos de edad de diagnóstico y grado de hiperpotasemia y/o acidosis metabólica. Se presenta una gran familia española compuesta de una niña y 5 adultos (total 6 pacientes), que mostraron una mutación del gen WNK1 (c.1889A>G, p.E630G). El diagnóstico clínico y de laboratorio se estableció por la presencia de hipertensión, hiperpotasemia, hipercloremia, acidosis metabólica, EFK <6%, EFNa <1%, EFCl <1,2%, renina inhibida, aldosterona normal y en algunos casos hipercalciuria (Ca/creatinina, g/g >0,2). En todos los casos la hipertensión y los trastornos electrolíticos y ácido-base se normalizaron inmediatamente con restricción de sal en la dieta y la administración de tiazida o indapamida a largo plazo.
ENFERMEDAD DE DENT TIPO 1 MANIFESTADA COMO PROTEINURIA Y GLOMERULOSCLEROSIS FOCAL: PRESENTACIÓN DE UNA FAMILIA CON UNA MUTACIÓN DEL GEN CLCN5
RP. RAMON PECES1, RM. ROCIO MENA2, CV. CRISTINA VEGA1, BR. BEGOÑA RIVAS1, CP. CARLOS PECES1, RS. RAFAEL SELGAS1, JN. JULIAN NEVADO2
1NEFROLOGÍA. HOSPITAL LA PAZ (MADRID), 2GENETICA. HOSPITAL LA PAZ (MADRID)
La enfermedad de Dent tipo 1 es una rara tubulopatía de herencia recesiva ligada al cromosoma X y caracterizada por proteinuria de bajo peso molecular, hipercalciuria, nefrocalcinosis, nefrolitiasis, hipofosfatemia y disfunción renal. Está causada por mutaciones del gen CLCN5 localizado en el cromosoma Xp11.22. El gen CLCN5 codifica el intercambiador electrogénico cloro/protones ClC-5 que está implicado en la reabsorción tubular of albúmina y proteínas de bajo peso molecular. Se presenta una gran familia española compuesta de varios miembros (4 varones enfermos y 4 mujeres portadoras) con enfermedad de Dent tipo 1 que presentaron distintos grados de albuminuria, proteinuria de bajo peso molecular (µ-albumina, α1-microglobulina), aminoaciduria, hipercalciuria, nefrocalcinosis, nefrolitiasis e hipofosfatemia asociados a glomerulosclerosis focal. Los varones presentaron distintos grados de insuficiencia renal precisando uno de ellos tratamiento renal sustitutivo. El análisis genético mostró una deleción frameshift en el exón 8 del gen CLCN5: c.653_654delCT (p.Leu218Profs*24). En algunos de los casos el tratamiento con fosfato, IECA, ARA II y espironolactona o tiazida mejoró las alteraciones urinarias.
ESTUDIO POBLACIONAL ACTUAL DE POLIQUISTOSIS RENAL AUTOSÓMICA DOMINANTE (PQAD) DEL ÁREA SANITARIA DE LEÓN Y LOCALIZACIÓN DE AQUELLOS PACIENTES SUBSIDARIOS DE RECIBIR TOLVAPTÁN
E. MONFÁ1, C. LUCAS1, J. GONZÁLEZ-ARREGOCÉS1, I. ROMANIOUK1, S. MARIÑO1, A. SASTRE1, B. DE LEÓN1, J. STEFAN1, M. PRIETO1
1NEFROLOGÍA. COMPLEJO ASISTENCIAL UNIVERSITARIO DE LEÓN (LEÓN)
Introducción: La PQAD es la enfermedad renal hereditaria más frecuente. Es la responsable del 7-10% de los pacientes en tratamiento renal sustitutivo (TRS) y por lo tanto, con un gran impacto social y económico. Aunque hay diversas estrategias para intentar enlentecer la progresión de ERC, Tolvaptán es el único tratamiento que ha mostrado un efecto beneficioso, logrando reducir la tasa anual de pérdida de función renal en un 30% frente a placebo (p<0,001). Recientemente, la Agencia Europea del Medicamento (AEM) ha aprobado su indicación para pacientes con PQAD. Nuestro objetivo es conocer la prevalencia en el área sanitaria de León y localizar aquellos pacientes subsidarios de recibir Tolvaptán.
Material y métodos: Los pacientes se han recogido en una base de datos prospectivamente actualizada junto con el registro de diálisis y trasplante de Castilla y León. El diagnóstico de los casos nuevos se ha realizado teniendo en cuenta los criterios de Ravine modificados.
Resultados: El número de pacientes vivos diagnosticados de PQAD y conocidos por nuestro servicio son 139 (0,8/2000 personas). El 33,1% están con TRS (19,4% trasplantados, 13,7% en diálisis); 5% en ERCA (eFG < 20 ml/min/1,73m2 -CKD-EPI); 61,9% en consulta externa. La indicación de Tolvaptán de la AEM es para pacientes con ERC estadio 1-3 con datos de progresión rápida (>2,5 ml/min/1,73m2 /año). De los prevalentes en nuestra consulta, 82,5% tienen un eFG ≥30 ml/min/1,73m2 -CKD-EPI; 36% son progresadores rápidos y el 29% conocerían ambas características. La edad media de inicio del tratamiento renal sustitutivo para pacientes con PQAD en nuestra población es de 44,66 +/- 14,57 años y el tiempo medio en TRS de 10 +/- 7,47 años.
Conclusiones: La prevalencia de pacientes con PQAD es inferior a la encontrada en otras zonas geográficas. Se podría explicar por varios motivos: menor afectación en nuestra población (enfermedad genética); emigración de la gente joven a otras ciudades con más oportunidades laborales: o desconocimiento de algunos pacientes afectos que no siguen revisiones en Nefrología por estar asintomáticos en las primeras etapas de la enfermedad.
El 29% de los pacientes con PQAD en consulta externa son subsidarios actualmente de recibir Tolvaptán, con el enlentecimiento de la progresión renal que ello supone, y el consecuente retraso en el inicio de TRS.

