










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNutrición Hospitalaria
On-line version ISSN 1699-5198Print version ISSN 0212-1611
Nutr. Hosp. vol.27 n.5 Madrid Sep./Oct. 2012
https://dx.doi.org/10.3305/nh.2012.27.5.5945
Importancia del diagnóstico precoz de fenilcetonuria en la mujer y del control de los niveles de fenilalanina en la gestación
Importance of early diagnosis of phenylketonuria in women and control of phenylalanine levels during pregnancy
F. Arrieta Blanco1,2, A. Bélanger Quintana1,3, C. Vázquez Martínez2 y M. Martínez Pardo1,3
1Unidad de Enfermedades Metabólicas/E raras. HU Ramón y Cajal. IRYCIS
2Departamento de Endocrinología y Nutrición (Unidad Nutrición, Obesidad y Metabolismo) HU Ramón y Cajal. IRYCIS. CIBEROBN
3Departamento de Pediatría. HU Ramón y Cajal. IRYCIS. CIBERER. Madrid. España
Dirección para correspondencia
RESUMEN
La Fenilalanina Hidroxilasa (PAH) hidroxila a nivel hepático la fenilalanina proveniente de la dieta. Los fetos dependen para la hidroxilación de fenilalanina de la función materna, ya que por inmadurez fetal esta función no se adquiere hasta la semana 26. Los pacientes con deficiencia de PAH (Fenilcetonuria, PKU) no hidroxilan adecuadamente la fenilalanina de la dieta por lo que sus niveles en sangre estan elevados. Los niveles de fenilalaninemia se consideran teratogénicos y neurotóxicos por encima de 360 umol/L (N < 120). Las mujeres PKU embarazadas deberán seguir estrictamente un tratamiento dietético y/o farmacológico para mantener niveles de fenilalaninemia < 180 umol/L y evitar las posibles complicaciones teratogénicas en el feto (Sindrome Fetal de Hiperfenilalaninemia Materna), como el caso que presentamos. Recomendamos descartar Fenilcetonuria en mujeres en quienes no se haya realizado un despistaje neonatal y/o tengan abortos, hijos con microcefalia, cardiopatía o malformaciones renales.
Palabras clave: Fenilcetonuria. Gestación. Dieta. Complicaciones.
ABSTRACT
The phenylalanine hydroxylase (PAH) in the liver hydroxylates phenylalanine from the diet. Fetuses depend for the hydroxylation of phenylalanine the maternal metabolism , fetal maturity does not come until week 26. Though the women with PAH deficiency (phenylketonuria, PKU) not adequately hydroxylate phenylalanine diet so their blood levels are high. Fenilalaninemia levels are considered neurotoxic teratogenic and above 360 umol/L (N < 120). Pregnant women should strictly follow PKU dietary treatment and/or drug to maintain levels of fenilalaninemia < 180 umol/L and avoid the teratogenic complications in the fetus (Hyperphenylalaninaemias Maternal Fetal Syndrome), as the case presented. We recommend discarding Phenylketonuria in women who have not been done a neonatal screening and/or have abortions, children with microcephaly, cardiac or renal malformations.
Key words: Phenilketonuria. Pregnant. Diet. Complications.
Introducción
La Fenilcetonuria (PKU) es una enfermedad de herencia autosómica recesiva en la que se afecta la hidroxilación hepática de fenilalanina (Phe), con defecto en la actividad Fenilalanina hidroxilasa (PAH) (fig. 1). En los pacientes PKU hay un aumento mantenido de Phe en sangre > 120 micromol/L que en el sistema nervioso central condicionan una apoptosis neuronal tanto mayor cuanto menos desarrollado este el cerebro1. Se consideran niveles adecuados para el desarrollo cerebral normal en niños aquellos < 360 umol/L, pero se pueden permitir niveles superiores en pacientes mayores. Los niveles de fenilalanina y condiciones personales y ambientales dan lugar a una afectación neurològica variable, desde la normalidad hasta un retraso mental grave, pudiendo pasar en algunos casos completamente desapercibido. El despistaje neonatal permite detectar la mayoría de los pacientes PKU en edades tempranas, pero aquellos con niveles muy bajos al nacer o en los que no se ha realizado este estudio pueden llegar a edades adultas sin diagnóstico.
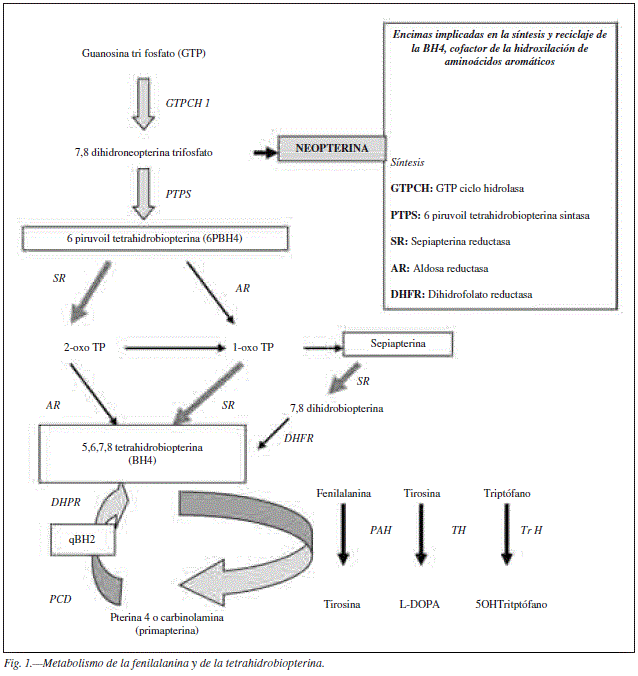
El Síndrome Fetal por Hiperfenilalaninemia Materna (SFM) se debe al efecto teratogénico de la fenilalanina en el feto durante el embarazo, aunque este aumento sólo se haya producido en las primeras semanas del embarazo. Este síndrome fue descrito por Dont en 1956, y se caracteriza clínicamente por microcefalia desde nacimiento con o sin retraso mental, abortos de repetición, cardiopatía (especialmente malformaciones de cavidades izquierdas) y malformaciones renales. El feto dobla los niveles de Phe maternos ya que la actividad PAH hepática del feto no está desarrollada hasta la 26 semana de embarazo y la madre es el único filtro metabólico para la fenilalanina. Dado que el efecto teratogénico y neurotóxico de la fenilalanina es de 360 umol/L, las mujeres embarazadas deben mantener niveles de fenilalanina en sangre por debajo de 180 umol/L para evitarlo. Las mujeres adultas PKU, incluso aquellas en tratamiento y bien controladas, tienen habitualmente niveles de fenilalanina en sangre entre 200 y 800 umol/L, es decir, superiores a los adecuados para evitar el riesgo de SFM. Por este motivo las mujeres PKU deben seguir un tratamiento estricto durante el embarazo que permita reducir dichos niveles a los adecuados para el correcto desarrollo fetal2,3.
En España, a muchas de las mujeres en edad fértil se les ha realizado el despistaje neonatal para PKU4. Sin embargo, sigue existiendo una población de riesgo para SFM. Se trata de aquellas mujeres nacidas antes del despistaje o provenientes de países donde no se realiza. El SFM debemos sospecharlo tanto si la madre tiene un diagnóstico previo de PKU como en toda madre con retraso mental de mayor o menor grado, inteligencia límite sin diagnóstico, y/o en aquellas que sin tener retraso mental, tengan o hayan tenido abortos de repetición, hijos previos con microcefalia desde el nacimiento con o sin retraso mental, cardiopatía y malformaciones renales.
En este trabajo describimos una paciente de 34 años de edad, con retraso mental leve, quien nos fue remitida para estudio, por haber tenido dos hijos de 12 y 9 años de edad con retraso mental y microcefalia y otro hijo fallecido por cardiopatía congénita.
Caso clínico
Mujer de 34 años de edad, de nacionalidad Argentina, remitida desde Neuropediatría a la Unidad de Enfermedades Metabólicas del Hospital Ramón y Cajal, con el fin de descartar enfermedad metabólica, por tener 2 hijos de 12 y 9 años, de diferente padre, con microcefalia y retraso mental severo. La paciente refería haber tenido otro hijo que, por malformación cardiaca, falleció a los 3 meses de edad. Estaba casada, hablaba normal y realizaba las actividades de la vida diaria. Tenía a veces problemas con el cambio del dinero y no había podido completar sus estudios pero sabía leer, escribir, sumar, restar, multiplicar y dividir (sólo por 1 cifra). Se le estimó un coeficiente intelectual de 70-75, correspondiente con un retraso mental leve-moderado. No realizaba ningún tratamiento ni refería enfermedades conocidas y no tenía antecedentes familiares de interés.
En ambos hijos los niveles de Phe fueron normales, de 70 y 92 micromol/L. Pero se determinaron los niveles de Phe en la paciente, siendo de 1.140 micromol/L (normal < 120 micromol/L). Los niveles de Phe al diagnóstico, entre 660 y 1.200 micromol/L condicionan una clasificación de fenotipo PKU suave-moderado. Se efectuó el diagnóstico diferencial con trastornos en el metabolismo de las pterinas, cuyo estudio fue normal. Se encontraron los siguientes cambios mutacionales en el gen PAH: c.165delT (p.Phe55fs) / c.q62G > A (p.Val388Met), siendo ambos hijos únicamente portadores de la mutación p.Phe55fs.
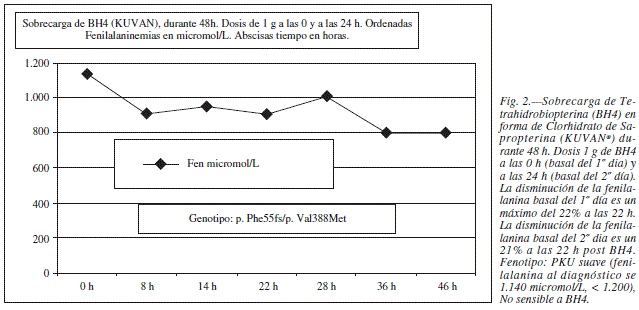
Tras el diagnostico de Fenilcetonuria en la madre se realizo una sobrecarga de Tetrahidrobiopterina (BH4) para descatar una posible respuesta a este tratamiento farmacológico, ya que en algunos pacientes PKU es posible mejorar la actividad PAH al dar dosis farmacológicas de su cofactor (fig. 2), pero en esta paciente los niveles de fenilalanina no experimentaron cambios significativos y por lo tanto se la consideró no respondedora.
Se inicio tratamiento con dieta limitada en fenilalanina con el fin de mantener niveles de fenilalanina menores de 660 micromol/L (niveles máximos permitidos en adultos sin embarazo) y control semanal de fenilalanina en sangre. Para ello ajustamos la alimentación a 20-25 g de proteínas de alto valor biológico/día (PAVB: leche, huevos, carnes, pescados, quesos, cereales y legumbres) repartidas en desayuno, comida y cena (5-10 y 10 g), con libertad en verduras, hortalizas, frutas naturales, patata pelada, aceitunas, azúcar, aceites, mantequilla, almidón de maíz (Maizena®), especias y alimentos de bajo contenido proteico Loprofín®, Aglutella® y Aproten® comprados a través de la Asociación de enfermos PKU de Madrid.
A las PAVB se le añadieron 60 g de proteínas especiales sin fenilalanina (PrXPhe) repartidas en 4 tomas/día, en forma de preparados especiales que contienen aminoácidos esenciales sin fenilalanina con hidratos de carbono, ácidos grasos esenciales, iones, oligoelementos y vitaminas.
La evolución semanal de las Phe a lo largo de los primeros 3 meses de seguimiento mostraron niveles de 498 ± 15 micromoles/L. Posteriormente la familia volvió a Argentina donde sigue tratamiento en la actualidad.
Discusión
La Phe es un aminoácido esencial e indispensable como nutriente. Se ingiere en forma de proteínas, se absorbe a nivel intestinal y se hidroxila en el hepatocito sintetizando tirosina, utilizando como cofactor la tetrahidrobiopterina (BH4) (fig. 1). Una parte que oscila entre el 10 y el 40% según las distintas etapas de crecimiento se utiliza para la síntesis proteica endógena1.
Los pacientes con mutaciones en el gen de la fenilalanina hidroxilasa, y dependiendo de la actividad residual de ésta, acumulan fenilalanina en sangre y otros tejidos. Para clasificar a los pacientes en distintos grupos de gravedad se tienen en cuenta las mutaciones, los niveles de fenilalanina al diagnóstico y cuánta fenilalanina son capaces de ingerir sin sobrepasar niveles de riesgo una vez inician el tratamiento (tolerancia)1.
La acumulación de fenilalanina actúa como tóxico directo, así como interfiriendo en el trasporte y metabolismo de otros aminoácidos neutros. A nivel del sistema nervioso central da lugar a distintos grados de afectación, llegando a un retraso mental profundo. En adultos que dejan la dieta se ha observado una regresión de sus capacidades mentales y la aparición de síntomas neuropsicológicos como depresión, ansiedad e incluso síntomas esquizoides. Por este motivo hoy en día se recomienda mantener el tratamiento de por vida1.
El tratamiento habitual de la fenilcetonuria consiste en una dieta limitada en fenilalanina, para lo cual se limita la ingesta de proteínas naturales de alto valor biológico (PAVB) en mayor o menor grado según la tolerancia del paciente. Aquellos con formas benignas podrán seguir una dieta normal (80-100 g PNVB/d), mientras que aquellos con las formas más graves no podrán sobrepasar los 2-6 g PAVB/d. La restricción en proteínas naturales debe compensarse con la ingesta de productos proteicos sin fenilalanina enriquecidas en tirosina, grasas, vitaminas y micronutrientes5,6. Las frutas y verduras se pueden administrar libremente7. Con esta dieta el desarrollo psicosomático es normal8.
En los últimos 10 años se ha observado que algunos pacientes PKU, sobre todo aquellos con formas suaves de la enfermedad, aumentan su tolerancia a proteínas al ser tratados con dosis farmacológicas de la tetrahidrobiopterina. Los pacientes respondedores pueden doblar o incluso normalizar su ingesta proteica9. A falta de estudios de seguridad, su uso no está autorizado durante el embarazo pero sus ventajas hacen que ya se utilice de forma esporádica en esta situación10.
Menos conocidos son los mecanismos teratogénicos de la fenilalanina. Hacer el diagnóstico es crucial para evitar que estas pacientes, como nuestro caso, tengan varios hijos afectos. A pesar de que muchas mujeres en edad fértil en nuestro país habrán sido estudiadas en periodo neonatal, siguen existiendo una importante cantidad de mujeres que se beneficiarían de realizar un despistaje de fenilcetonuria en edad adulta.
Conclusión
La determinación de fenilalanina en sangre en embarazadas y en mujeres fértiles a quienes no se le haya efectuado el cribado neonatal, debe ser obligatoria para detectar mujeres PKU, cuyos hijos pueden beneficiarse de un tratamiento dietético efectuado a la madre durante el embarazo.
Referencias
1. Scriver CR, Kaufman S, Eisensmith RC, Woo SLC: The hyperphenylalaninemias. In The metabolic and molecular bases of inherited disease. 7th edition. Edited by Scriver CR, Beaudet AL, Sly WS, Valle D. McGraw-Hill, New York; 1995: 1015-75. [ Links ]
2. Prick BW, Hop WC, Duvekot JJ. Maternal phenylketonuria and hyperphenylalaninemia in pregnancy: pregnancy complictions and neonatal sequelae in untreated and treated pregnancies. Am J Clin Nutr 2012; 95: 374-82 [ Links ]
3. Hanley WB, Azen C, Koch R, Michals-Matalon K, Matalon R, Rouse B, Trefz F, Waisbren S, de la Cruz F. Maternal Phenylketonuria Collaborative Study (MPKUCS)-the "outliers". J Inherit Metab Dis 2004; 27 (6): 711-23. [ Links ]
4. Dulín E, Cortés E, Chamorro F, Eguileor I, Espada M, Pámpols T et al. Estado actual de los programas de cribado neonatal en España. Acta Pediatr Esp 2001; 59: 467-78. [ Links ]
5. Blau N, van Spronsen FJ, Levy HL: Phenylketonuria. Lancet 2010; 376: 1417-27. [ Links ]
6. MacDonald A, Rocha JC, van Rijn M, Feillet F. Nutrition in phenylketonuria. Mol Genet Metab 2011; 104 (Suppl.): S10-8. [ Links ]
7. Martínez-Pardo M, Bélanger-Quintana A, García Muñoz MJ, Desviat L, Pérez B, Ugarte M. Fenilcetonuria. Protocolo de Diagnóstico, tratamiento y seguimiento de las hiperfenilalaninemias. Libro: Protocolos de diagnóstico y tratamiento de los Errores Congénitos del Metabolismo. Protocolos oficiales de la AECOM. Editor: P. Sanjurjo. Ediciones Mead-Johnson. 2007. [ Links ]
8. MacDonald A, Rylance G, Davies P, Asplin D, Hall SK, Booth IW. Free use of fruits and vegetables in phenylketonuria. J Inherit Metab Dis 2003; 26 (4): 327-38. [ Links ]
9. Bélanger-Quintana A, García MJ, Castro M, Desviat LR, Pérez B, Mejía B, Ugarte M, Martínez-Pardo M Spanish BH4-responsive phenylalanine hydroxylase-deficient patients: evolution of seven patients on long-term treatment with tetrahydrobiopterin. Mol Genet Metab 2005; 86: S61-6. [ Links ]
10. Belanger-Quintana A, Martínez-Pardo M. Physical development in patients with phenylketonuria on dietary treatment: a retrospective study. Mol Genet Metab 2011; 104: 480-4. [ Links ]
11. Trefz FK, Belanger-Quintana A Sapropterin dihydrochloride: a new drug and a new concept in the management of phenylketonuria. Drugs Today (Bare) 2010; 46: 589-600. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Francisco Arrieta Blanco
Hospital Ramon y Cajal
C/ Colmenar km 7,2
Madrid. España
E-mail: arri68@hotmail.com
Recibido: 3-V-2012
Aceptado: 20-VI-2012

