










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales de Medicina Interna
Print version ISSN 0212-7199
An. Med. Interna (Madrid) vol.24 n.9 Sep. 2007
Síndrome pluriglandular autoinmune. Revisión
Pluriglandular autoimmune syndrome. Systematic review
M. J. Molina Garrido, C. Guillén Ponce, M. Guirado Risueño, A. Mora1, A. Carrato
Servicios de Oncología Médica y 1Medicina Interna. Hospital General Universitario. Elche. Alicante
Dirección para correspondencia
RESUMEN
Se han descrito múltiples casos de síndromes autoinmunes que cursan con hipofunción glandular asociada a otras enfermedades, no siempre endocrinológicas, que, en ocasiones, presentan agregación familiar. Se conoce como síndromes poliglandulares autoinmunes (SPA) aquellos en los que coexisten al menos dos insuficiencias de glándulas endocrinas como consecuencia de un mecanismo autoinmune basado en la acción de los autoanticuerpos o los linfocitos T activados frente a distintos antígenos de los órganos diana o de las glándulas endocrinas, o en el bloqueo de la función hormonal. A continuación, se presenta una revisión de los principales síndromes autoinmunes, los criterios diagnósticos de los mismos y los procesos autoinmunes involucrados.
Palabras clave: Autoinmunidad. Síndrome pluriglandular. Insuficiencia suprarrenal autoinmune.
ABSTRACT
There are a lot of autoimmune syndromes with glandular disfunction which are associated to another diseases. Sometimes, these processes are associated to similar cases in the same family. Autoimmune polyglandular syndromes are characterized by the coexistence of two or more endocrine insufficiencies due to an autoimmune mechanism: the activity of autoantibodies or T activated lymphocytes against organs or endocrine glands. In this report, they have been described the main autoimmune syndromes, the diagnostic methods and the autoimmune mechanisms which take a role in their origin.
Key words: Autoimmunity. Pluriglandular syndrome. Autoimmune adrenal insufficiency.
Introducción
En 1849 Addison describió la asociación de anemia perniciosa con insuficiencia suprarrenal primaria, y es a partir de este momento cuando se describe un sinfín de enfermedades de base autoinmune. Schmidt, en 1926, describió la necropsia de dos pacientes que murieron con insuficiencia suprarrenal, en los que se apreciaba una infiltración linfocitaria destructiva en las glándulas tiroides y suprarrenales. Él lo denominó "eine Biglandulare Erkrankung" (enfermedad de dos glándulas). En 1929 se describió la primera asociación entre el hipoparatiroidismo y la candidiasis mucocutánea. En 1946 se comunicó la asociación de estas dos enfermedades con la insuficiencia suprarrenal idiopática. En 1980 se introdujo el término de "síndrome pluriglandular autoinmunitario" para describir la asociación de insuficiencia suprarrenal primaria, tiroidopatía autoinmunitaria y candidiasis crónica cutaneomucosa (1). Desde este momento, quedaron definidos dos tipos fundamentales de síndromes poliglandulares autoinmunes: el tipo 1 asociaba la enfermedad de Addison con el hipoparatiroidismo, y el tipo 2, la insuficiencia suprarrenal con la diabetes mellitus tipo 1 y con la enfermedad tiroidea. Hay descritas otras asociaciones menos frecuentes, como la insuficiencia gonadal primaria (ovario o testículo), la insuficiencia hipofisaria, y otras enfermedades ya incluidas en el SPA tipo 1 y el tipo 2. Incluso se puede asociar otras enfermedades no endocrinas, aunque también de naturaleza autoinmune, como son la anemia perniciosa, la alopecia, malabsorción, hepatitis crónica activa y candidiasis cutaneomucosa. Recientemente se ha descrito un SPA tipo 3, en adultos, en el que, a diferencia de lo que ocurre en los tipos 1 y 2, no está afectada la corteza suprarrenal. Aparte de esta diferencia, no se ha descrito ninguna otra distinción entre los tipos 2 y 3 (2).
Etiopatogenia
El SPA es la causa más frecuente de infiltración linfocitaria de las glándulas endocrinas con destrucción progresiva y lenta de las mismas, que acaba causando insuficiencia hormonal. Esto implica que en el SPA no siempre exista manifestaciones clínicas de cada una de las enfermedades del síndrome (todo depende del grado y la velocidad de destrucción de la glándula). Existe, por tanto, un periodo preclínico en el que se detecta los anticuerpos circulantes frente a la célula diana, sin que exista destrucción glandular total, y un periodo posterior en el que puede producirse la insuficiencia glandular. Esto hace posible el diagnóstico de un SPA cuando se haya manifestado sólo una de las enfermedades endocrinas; por tanto, la detección de autoanticuerpos en fases en las que aún no haya una endocrinopatía manifiesta permite establecer un diagnóstico precoz del SPA. La prevalencia de estos anticuerpos varía; algunos se detectan en sangre de pacientes que nunca presentarán manifestaciones clínicas del síndrome pluriglandular (esto ocurre cuando la destrucción de la glándula endocrina es muy lenta). Aunque la pérdida de masa glandular no suele ser rápida en las enfermedades autoinmunitarias, en los paciente jóvenes, un factor externo, como una infección, puede desencadenar una de las enfermedades asociadas de forma brusca. Hay distintos datos que indican que se trata de un proceso autoinmune: en los órganos afectos existe un infiltrado inflamatorio crónico de predominio linfocitario; en los del tipo 2 hay una estrecha relación con HLA-B8; se ha detectado autoanticuerpos circulantes, que reaccionan contra antígenos específicos de órganos, como son moléculas de superficie (receptores), enzimas intracelulares, proteínas secretadas u hormonas segregadas por el órgano (3). Incluso se han descrito casos de asociación entre endocrinopatías y otros síndromes también autoinmunitarios, como la inmunodeficiencia variable común. En estos casos, el origen de ambos procesos es autoinmune (4).
Los SPA se producen como consecuencia de la pérdida de inmunotolerancia frente a las propias proteínas que actúan como antígenos. No se conoce bien el motivo de este suceso, pero se postula una serie de hipótesis: la pérdida de la capacidad de supresión de las clonas autorreactivas, la presencia de antígenos extraños al organismo, o la liberación de antígenos habitualmente no accesibles al sistema inmunitario. Algunos de los anticuerpos más descritos en el SPA aparecen reflejados en tabla I (2).
Presentación clínica
Normalmente, la presentación clínica de los SPA suele ocurrir por etapas. En la mayoría de los casos se manifiesta una enfermedad endocrina de etiología autoinmunitaria, y, tras unos meses o incluso años, van apareciendo otras, que no siempre son endocrinas, con la que llega a establecerse una relación (1).
Ante la manifestación clínica de una enfermedad endocrina autoinmune, debe sospecharse, o, al menos, descartarse, que se trate de un SPA. Éste puede descubrirse hasta en un 50% de los pacientes afectos por la enfermedad de Addison. Normalmente, en los pacientes jóvenes, el diagnóstico de diabetes mellitus precede al de la enfermedad de Addison, aunque la tiroidopatía autoinmune puede precederla o sucederla (5). Los marcadores más tempranos del SPA son los autoanticuerpos frente al tejido glandular (anticuerpos antiislote pancreático, antimicrosoma, antisuprarrenales), al producto de secreción o las proteínas transportadoras del mismo (insulina, tiroglobulina). En muchos casos, la secreción hormonal basal es normal, en especial, en las fases tempranas de la enfermedad, por lo que las determinaciones hormonales no sólo deben hacerse en condiciones basales, sino también tras un estímulo del factor liberador hormonal. En muchas ocasiones en las que la secreción basal es normal, pueden hallarse respuestas abolidas o escasas frente al estímulo. En estos casos, incluso la clínica puede ser totalmente insidiosa y no manifestarse (1).
Clasificación de los SPA
Inicialmente, se clasificó los SPA en dos grupos, los tipos 1 y 2. Con el tiempo, se ha ido definiendo otros síndromes con un origen autoinmune demostrado, que no cumplían totalmente las condiciones establecidas en la clasificación previa. Betterle et al en el año 2002, publicaron una nueva clasificación, basada en Neufeld y Blizzard (2), que aún está vigente en nuestros días (Fig. 1). Según esta clasificación: SPA tipo 1: debe cumplir, al menos, dos de estas condiciones: Candidiasis crónica, hipoparatiroidismo crónico, insuficiencia suprarrenal autoinmune. SPA tipo 2: insuficiencia suprarrenal (requisito indispensable), enfermedad tiroidea autoinmune (tiroiditis de Hashimoto o enfermedad de Graves) y/o diabetes mellitus tipo 1. Es condición obligatoria que esté presente la insuficiencia suprarrenal. SPA tipo 3: enfermedad tiroidea autoinmune y otras enfermedades autoinmunes, exceptuando la insuficiencia suprarrenal autoinmune, el hipoparatiroidismo o la candidiasis crónica. Debe existir, al menos, una de las siguientes enfermedades autoinmunes: vitíligo, alopecia, anemia perniciosa o diabetes mellitus tipo I. Es indistinguible del tipo 2, salvo por la insuficiencia suprarrenal, un criterio que, en opinión de Muir y cols., es arbitrario (6). SPA tipo 4: 2 o más enfermedades autoinmunes específicas de órgano que no cumpla criterios de tipo 1, 2 ni 3.

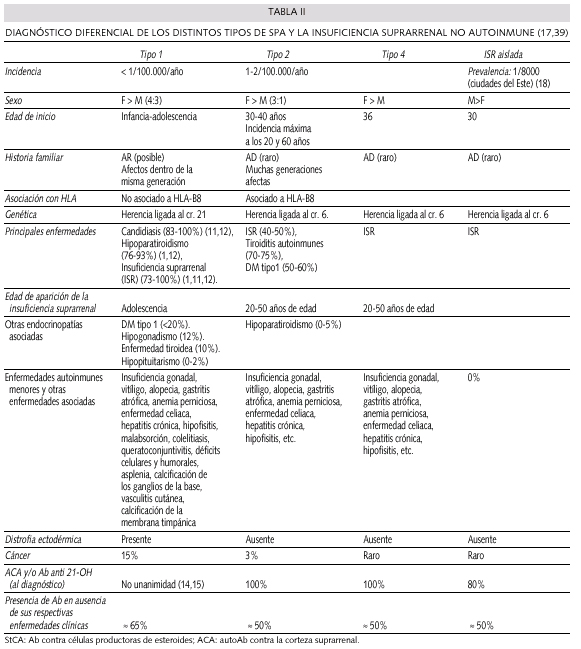
En la tabla II se resumen las principales características de los SPA, así como los tipos descritos hasta el momento y algunos datos diferenciales con la insuficiencia suprarrenal primaria.
TIPO 1
Este síndrome también es conocido como APECED (autoimmune polyendocrinopathy, candidiasis and ectodermal dystrophy) o MEDAC (multiple endocrine deficiencyt autoimmune candidiasis syndrome). Se ha empleado otros nombres como el del síndrome de Whitaker (7). La herencia es autosómica recesiva y está asociada al gen 21q22.3, con la mutación R257X como responsable del 82% de los casos (8,9).
Por lo general, la primera manifestación suele aparecer entre los 3-5 años de edad, o en los primeros años de la adolescencia, por lo que también se denomina poliendocrinopatía autoinmune juvenil (10). Los tres principales componentes del síndrome suelen aparecer antes de los 20 años, mientras que el resto de enfermedades autoinmunes pueden manifestarse incluso en la quinta década de vida (11,12). En Finlandia, la prevalencia es próxima a 1/25.000 habitantes (1). La mayor prevalencia de esta rara enfermedad se ha descrito en poblaciones con un alto grado de consanguineidad o que son descendientes a su vez de pequeñas poblaciones, en especial, en los judíos de Irán (entre 1:600 a 1:9000) (7). Por el contrario, en otras poblaciones, la incidencia es mucho menor, como ocurre en Noruega (1:80000) (13). La relación mujer/hombre oscila entre 0,8/1 a 2,4/1, según las series (11,12).
Ya en 1929, Torpe y Hendley, describieron la asociación de candidiasis mucocutánea con insuficiencia glandular. En la actualidad, el síndrome pluriglandular autoinmune tipo 1 está definido por la presencia de insuficiencia suprarrenal asociada a una candidiasis mucocutánea crónica e hipoparatiroidismo. Estos tres componentes aparecen en un preciso orden cronológico, pero los tres, de forma conjunta, sólo están presentes en el 33-50% de estos pacientes (10,14). Se sabe que, cuanto menor sea la edad de aparición del primero de estos componentes, con mayor facilidad se desarrollará el resto, mientras que aquellos pacientes en los que las manifestaciones clínicas aparezcan más tardíamente, llegan a presentar menos componentes clínicos (1).
Este síndrome no se asocia con el HLA, ni a nivel demográfico, ni en las familias afectas. Suele limitarse a una generación de familiares, pudiendo incluso enfermar muchos de los miembros de la misma, pero no se afecta la generación posterior. Dentro de una misma generación, los pacientes pueden presentar sólo una de las tres enfermedades descritas.
Además de la candidiasis mucocutánea, el hipoparatiroidismo primario o la insuficiencia suprarrenal primaria, hay otras manifestaciones asociadas, que se reflejan en la tabla III. También existe predisposición a determinados tumores: carcinoma epitelial de la mucosa oral y del esófago, y adenocarcinoma de estómago.
La candidiasis mucocutánea, que suele afectar a piel, uñas y mucosa (bucal y anal), es la primera manifestación de este síndrome. Normalmente aparece antes de los 5 años de edad, aunque se ha descrito casos de aparición en el primer mes de nacimiento o incluso los 21 años de edad (11,15). En la serie de Betterle et al, la edad mediana de aparición fue de 6,5 años (12). Por este motivo, todos los pacientes afectos por candidiasis mucocutánea crónica, en especial, si son niños, deben ser evaluados periódicamente desde el punto de vista inmunológico, bioquímico, e incluso con tests clínicos, para reconocer la sintomatología de una insuficiencia glandular endocrina asociada (12). Existe algún caso aislado en el que se llegó al diagnóstico de SPA tipo 1 sin que hubiera aparecido la candidiasis mucocutánea (16).
Se considera que es una expresión clínica de un déficit inmunológico selectivo de células T ante Candida albicans, combinado con una respuesta normal de las células B frente a los antígenos de generados contra ésta, lo que previene el desarrollo de una candidiasis sistémica (12,21). Por este motivo, el SPA tipo 1 también ha sido considerado una inmunodeficiencia (22).
Afecta a la dermis y a la mucosa oral, vaginal y esofágica. En la mayoría de los casos, la afectación no supera el 5% de la superficie corporal (11). En algunos pacientes, la candidiasis conduce a la aparición de determinadas complicaciones, como dolor retroesternal o incluso estenosis esofágica (11,12). En algunos pacientes, incluso puede inducir el desarrollo de un carcinoma epitelial de la mucosa oral (14). A menudo es necesario un tratamiento antifúngico periódico según el protocolo de DePadova-Elder y cols. (23), aunque no suele dar buen resultado en las infecciones de mucosas (12).
Desde el diagnóstico de una patología hasta el diagnóstico de la siguiente, pueden pasar décadas. Suelen presentar alteración de la hipersensibilidad retardada, y se han descrito anormalidades de los linfocitos T. En los adultos, las candidiasis rara vez forman parte de un síndrome pluriglandular tipo 1, pero pueden tener relación con anormalidades inmunológicas que acompañan a los timomas.
El hipoparatiroidismo es la primera anomalía endocrinológica que aparece en los pacientes afectos del SPA tipo 1 (11). Suele desarrollarse antes de los 10 años de edad, oscilando entre los 3 meses desde el nacimiento hasta los 44 años de edad. En la serie de Betterle y cols., la mediana de edad de aparición fue de 9,2 años (12). Afecta al 73-100% de los pacientes (1,11,12). Cuando aparece durante el periodo neonatal, es importante diferenciarlo de enfermedades genéticas como el síndrome Di George (delección 22q11), caracterizado por déficits congénitos cardiacos, con afectación de grandes vasos, tetania por disminución del calcio sérico por fallo en el desarrollo del tejido paratiroideo, y ausencia de timo normal, lo que lleva a un defecto aislado de células T. Suele manifestarse de forma severa, con estridor laríngeo y espasmo carpopedal. En las autopsias realizadas a los pacientes con SPA tipo 1 e hipoparatiroidismo asociado, las glándulas paratiroides se mostraban atróficas. Li y cols. han detectado la presencia de autoanticuerpos contra el dominio extracelular de los receptores del calcio hasta en el 56% de los pacientes con hipoparatiroidismo adquirido, y la mayoría de estos pacientes tenía SPA tipo 1 (24).
La última de estas 3 manifestaciones es la insuficiencia suprarrenal primaria, que aparece entre los 6 meses y los 40 años de edad, con un pico alrededor de los 13 años (11), con clínica idéntica a los de cualquier insuficiencia adrenal aislada. En la serie de Betterle y cols. la mediana de edad de aparición fue de 9,2 años (12). Afecta al 70-100% de los casos, según las series de Neufeld, Ahonen, y Betterle (1,11,12). En las autopsias de estos pacientes, el tejido tisular se mostraba atrófico, con una distorsión que afectaba a la totalidad de la estructura de la corteza, y con nidos de fibrosis asociados. En algunos casos, también la médula estaba atrófica. En la actualidad, aunque no existe un consenso total acerca de los principales autoantígenos en el SPA tipo 1, sí que existe unanimidad en cuanto a que el principal autoantígeno es la 21-hidroxilasa, tanto en el SPA tipo 2 como en la insuficiencia suprarrenal aislada (25).
La insuficiencia gonadal es más frecuente en la mujer que en el hombre, y suele manifestarse con irregularidades en la menstruación.
Aunque puede aparecer antes de los 40 años (amenorrea secundaria), en ocasiones lo hace antes de la pubertad (amenorrea primaria) (26). Desde el punto de vista microscópico, el tejido gonadal muestra hipoplasia e infiltración linfocítica (25). En todos los casos se ha detectado anticuerpos contra las células productoras de esteroides. La presencia de este tipo de anticuerpos en mujeres con SPA tipo 1 sin hipogonadismo es un indicador precoz de la aparición posterior de hipogonadismo hipergonadotrópico (27).
TIPO 2 (SD. SCHMIDT)
El SPA tipo 2 es el más frecuente de los SPA descritos. Se estima que su prevalencia es de 1,4-2:100000 habitantes, y las mujeres se afectan unas 3 veces más que los hombres. Suele ocurrir en la edad adulta, entre la tercera y la cuarta décadas, y es muy raro que aparezca en la infancia (28). A diferencia de lo que ocurre en SPA tipo 1, los familiares suelen estar afectos con frecuencia (10). Se hereda con carácter dominante, lo que hace necesario que se establezca un protocolo de seguimiento en los pacientes con SPA tipo 2. En una familia en la que se haya documentado un caso de SPA tipo 1, hay que informar a los pacientes de los síntomas y los signos de las principales enfermedades que forman parte del síndrome. Aun en ausencia de sintomatología alguna, se debe examinar cada 3 ó 5 años a los pacientes con riesgo entre 20 y 60 años de edad, mediante la determinación de glucosa en ayunas, anticuerpos citoplasmáticos contra las células de los islotes, TSH, tiroxina en sangre, niveles de cortisol tras estimulación, junto con una anamnesis y un examen físico completos. Se debe recordar que pueden transcurrir incluso 20 años desde el diagnóstico de una endocrinopatía y la aparición de otra acompañante (29).
Se define por la concurrencia en un mismo paciente, de dos o más de los siguientes trastornos: insuficiencia suprarrenal primaria (enfermedad de Addison), hipertiroidismo o hipotiroidismo primarios autoinmunes (síndrome de Schmidt), diabetes mellitus tipo 1 (síndrome de Carpenter), hipogonadismo primario, miastenia grave y enfermedad celiaca. La anemia perniciosa, la alopecia, la insuficiencia gonadal y el vitíligo, ocurren con mayor frecuencia que en el SPA tipo 1. La insuficiencia suprarrenal puede preceder a las otras endocrinopatías (30). Suele aparecer entre los 20 y los 50 años de edad (1), y afecta más a mujeres que a hombres, con una relación 2:1 (31). Este predominio femenino es aún más claro cuando coexiste tiroiditis que cuamdo sólo existe una enfermedad de Addison o diabetes tipo 1. por lo tanto, el predominio de mujeres con enfermedad de Addison puede reflejar en la actualidad la influencia poderosa de los genes de la tiroiditis. También parece existir un predominio femenino en relación entre la autoinmunidad suprarrenal y la gonadal. Entre mujeres con ooforitis linfocítica demostrada mediante biopsia, casi siempre se produce insuficiencia suprarrenal o autoinmunidad suprarrenal subclínica (31). Las mujeres con enfermedad de Addison es muy raro que presenten una insuficiencia gonadal, aun incluso estando presentes autoanticuerpos gonadales.
Por orden de prevalencia decreciente, las distintas enfermedades que forman parte del SPA tipo 2 son: hipertiroidismo, tiroiditis atrófica, diabetes mellitus tipo 1, insuficiencia suprarrenal, enfermedad celiaca y miastenia gravis (2).
De entre todas las patologías que constituyen el SPA tipo 2, la tiroiditis autoinmune es el que con más frecuencia aparece de forma aislada. En la población general, su prevalencia es de 800 por cada 100.000 habitantes, con un importante predominio femenino (83-95%) (31). Su mayor incidencia coincide con dos picos: en la adolescencia, y entre los 50 y 60 años. Es muy raro que ocurra a edades más tempranas. En un estudio que englobaba a población japonesa con tiroiditis, el 7,6% tenía también autoanticuerpos contra las células de los islotes pancreáticos, y la gran mayoría de estos (20 de un total de 24) presentaba diabetes tipo 1 (32). Sólo el 1% de un total de 4.353 pacientes adultos con evidencia clínica de tiroiditis aislada tenían evidencia serológica de autoinmunidad suprarrenal (29). Normalmente, cuando existe patología tiroidea autoinmune, la afectación poliglandular es poco frecuente, y cuando esto ocurre, el resto de patologías suelen preceder al diagnóstico de la tiroiditis.
Se cree que existe una predisposición genética (relación con HLA), que, estimulada por algún factor ambiental, llega a desencadenar un fenómeno autoinmune que causa la destrucción o la hiperfunción glandular. No llega a heredarse la enfermedad específica en sí, sino la susceptibilidad a padecer el SPA. Aunque la mayoría de casos se asocian a HLA-B8, dicha asociación no suele existir en la anemia perniciosa, la tiroiditis bociosa ni el vitíligo (2).
En todos estos trastornos se detectan autoanticuerpos organoespecíficos. Algunos de estos anticuerpos son marcadores específicos de la enfermedad, e incluso participan directamente en la fisiopatología de la anomalía (Ab antiacetilcolina); en algunas ocasiones, los anticuerpos se detectan sin que haya enfermedad clínicamente detectable, o preceden, por años, a las manifestaciones de la enfermedad (anticuerpos antitiroglobulina, anticuerpos antimicrosomales tiroideos y anticuerpos anticélulas parietales); estos, suelen ser comunes también a los familiares sanos de los pacientes afectos. Otros Ab que están involucrados en la patología endocrina autoinmune son las inmunoglobulinas tiroestimulantes del hipertiroidismo, los Ab antimelanocíticos, los Ab antisuprarrenales y los Ab antigonadales (2).
En todos los pacientes con insuficiencia suprarrenal idiopática, se debe determinar TSH, tiroxina y glucemia basal, ya que en más del 45% de estos casos llega a desarrollarse otra u otras endocrinopatías. También debe investigarse la presencia de síntomas o signos de anemia perniciosa o de hipogonadismo, que suelen pasar inadvertidos. Si el paciente presenta una tiroideopatía aislada, sin antecedentes familiares de SPA tipo 2, no es necesario continuar con ningún tipo de evaluación endocrinológica acompañante, porque la incidencia de SPA en estos casos es muy baja.
TIPO 3
Inicialmente, este síndrome fue clasificado por Neufeld y Blizzard como la asociación entre una enfermedad tiroidea autoinmune y una o más enfermedad autoinmune. En función al tipo de enfermedad autoinmune asociada, se diferencian varios subtipos: diabetes mellitus tipo 1 en el SPA tipo 3 a; la gastritis atrófica, o anemia perniciosa en el tipo 3b y el vitíligo, alopecia o miastenia gravis en el tipo 3c. En la clasificación inicial, tanto la insuficiencia suprarrenal como el hipoparatiroidismo se consideraban como excluyentes. Las combinaciones posibles son tan variadas, que realmente se podría definir una infinitud de subtipos de SPA tipo 3 (2).
TIPO 4
El SPA tipo 4 es un síndrome raro que se caracteriza por la asociación de distintas combinaciones de enfermedades autoinmunes que no corresponda ninguno de los síndromes citados previamente. A los pacientes con SPA tipo 4 se les debe determinar anticuerpos antitiroideos, anticuerpos contra los islotes pancreáticos y anticuerpos contra la decarboxilasa del ácido glutámico, ya que la detección de alguno de estos permitiría diferenciar un falso SPA tipo 4 de un SPA tipo 2 latente. En estos pacientes, al igual que ocurre en los pacientes con SPA tipo 1 o tipo 2, la resonancia magnética revela atrofia de las glándulas suprarrenales (2).
Diagnóstico
En el momento en que se diagnostique una enfermedad glandular, debe descartarse la coexistencia con otros procesos. Se ha de hacer un screening con autoanticuerpos específicos para cada patología (17)
Tras llevar a cabo dicho screening, en función del órgano afecto, se debe efectuar un estudio exhaustivo de la función del órgano blanco, siguiendo los mismos pasos que se seguirían si el paciente presentara una patología endocrinológica primaria aislada. Para evaluar a los portadores de autoanticuerpos positivos, se puede determinar periódicamente los niveles de TSH, la calcemia total, el calcio iónico, la fosfatemia, glucemias basales y PTG. Si se detecta niveles elevados de gonadotropinas con valores bajos de 17-beta-estradiol o testosterona, existe una insuficiencia gonadal primaria asociada (17).
La presencia de autoanticuerpos no siempre es sinónimo de enfermedad o de predicción de que ésta vaya a producirse tras cierto periodo de tiempo. Algunos autoanticuerpos fluctúan, y están presentes sólo durante ciertos periodos de tiempo, lo que indica una agresión inmune autolimitada, algunas de estas enfermedades cursan en brotes, con periodos de actividad inmunitaria y periodos silentes, lo que se acompaña o no de la detección de anticuerpos en sangre (33). La detección de anticuerpos, por tanto, requiere un estudio hormonal para establecer el diagnóstico de certeza de SPA en estadio preclínico, que es el más difícil de detectar. Como resumen al screening, se puede decir que, a los pacientes con una endocrinopatía autoinmune uniglandular está recomendado hacerles un screening funcional para APS cada 3 años hasta los 75 años de edad (17). Si se detecta algún hallazgo patológico, como la aparición de una segunda enfermedad endocrinológica autoinmune, además, se debe hacer una medición de los autoanticuerpos específicos de órgano, e incluso un screening funcional de endocrinopatías autoinmunes en los familiares de primer grado en aquellos pacientes con un diagnóstico reciente de APS, el screening genético sólo sería útil en los APS tipo 1 (8).
Siempre es necesario explorar la función adrenal con la estimulación con ACTH para poner de manifiesto los estados subclínicos, pues el descenso del cortisol en sangre y las alteraciones electrolíticas suelen producirse cuando la insuficiencia suprarrenal ya está establecida totalmente (34).
En el diagnóstico, el médico debe ser muy prudente: el diagnóstico de SPA no debe pasar desapercibido, porque un diagnóstico temprano permite un tratamiento sustitutivo en las fases en las que aún no hay alteraciones importantes a nivel hidroelectrólitico ni a otros niveles. Por otro lado, no se debe dar un diagnóstico de SPA de forma precipitada, en el momento en que se haya detectado anticuerpos circulantes, puesto que la detección de los mismos no es un marcador fiable de enfermedad. La actitud más adecuada consistiría en hacer un control regular de los marcadores hormonales (valores normales y tras la estimulación) y de los marcadores inmunológicos (anticuerpos circulantes) en todos los pacientes con una endocrinopatía inmunitaria, para diagnosticar precozmente un SPA (33).
Tratamiento
El éxito en el manejo de los pacientes con SPA radica en detectar pronto la patología y tratarla antes de que cause una morbimortalidad importante. El tratamiento de las distintas insuficiencias glandulares es igual al que se administraría ante una patología idéntica que apareciera de forma primaria y aislada. El uso de tiroxina puede desencadenar una crisis suprarrenal, que puede incluso conducir a la muerte a los pacientes con insuficiencia suprarrenal no tratada e hipotiroidismo: en todos los pacientes hipotiroideos en los que se sospeche este síndrome, antes de iniciar el tratamiento con tiroxina se debe evaluar la función suprarrenal (35,36). En aquellos pacientes con insuficiencia suprarrenal e hipotiroidismo primario, la función tiroidea mejora tras el tratamiento sustitutivo con glucocorticoides.
El tratamiento de la insuficiencia suprarrenal sintomática se basa en el uso de hidrocortisona o cortisona por la mañana y en la tarde. La dosis inicial aconsejada es de 25 mg de hidrocortisona (dividida en dosis de 15 y 10 mg) o 37,5 mg de cortisona (dividida en dosis de 25 y 12,5 mg), pero la dosis diaria puede ser reducida a 20 ó 15 mg de hidrocortisona en el momento en que el paciente se encuentre bien. Con el fin de prevenir el aumento de peso y la osteoporosis, la norma a seguir debe ser emplear la dosis más pequeña de corticoides que permita controlar la sintomatología clínica. Con la finalidad de conocer la dosis adecuada de hidrocortisona, puede ser útil determinar los niveles de cortisol en orina (33). Los pacientes con insuficiencia suprarrenal primaria también deberían recibir fludrocortisona, a dosis diarias de 50-200 microgramos, como sustituto de la aldosterona. La dosis adecuada se determinará con la medida de la presión arterial sanguínea y del potasio en sangre, que deben mantenerse en niveles medios-altos. Todos los pacientes con insuficiencia suprarrenal debieran llevar una tarjeta identificativa en la que se indique el tratamiento actual y las recomendaciones terapéuticas en el caso de que empeoren clínicamente. Estos pacientes, deben aumentar la dosis de hidrocortisona al doble o al triple temporalmente cuando tengan fiebre o cuando sufran cualquier lesión, como ocurre en las intervenciones quirúrgicas (37).
El tratamiento menos efectivo es el de la diabetes mellitus, pues, a pesar de éste, suelen aparecer complicaciones vasculares de forma tardía. La prednisona no parece modificar la evolución clínica si se administra una vez que se ha producido una diabetes franca. A este respecto, existe controversia sobre el uso de ciclosporina A y de azatioprina. Ninguno de estos fármacos, empleados en ensayos, produjo una respuesta clínica lo suficientemente prolongada como para justificar su uso, teniendo en cuenta su toxicidad potencial. Los efectos secundarios de la ciclosporina son la nefrotoxicidad, la hepatotoxicidad, el hirsutismo, hiperplasia gingival, descenso de los valores de hemoglobina, y tendencia a la aparición de linfomas. La azatioprina puede producir mielosupresión y neoplasias malignas. Todos estos efectos secundarios, así como los resultados obtenidos en los estudios llevados a cabo, sugieren que no debe emplearse el tratamiento inmunosupresor fuera de ensayos clínicos, totalmente monitorizados.
En los pacientes con diabetes mellitus en los que cada vez sea necesaria una menor dosis de insulina, se debe sospechar el inicio de una insuficiencia suprarrenal en la que aún no han aparecido hiperpigmentación ni alteraciones electrolíticas (33).
La candidiasis mucocutánea se puede tratar con ketoconazol (23), que puede inducir una mayor inhibición en la síntesis de cortisol. En la mayoría de estos pacientes, la enfermedad recidiva al suspender el fármaco o al reducir sus dosis, e incluso se han descrito recaídas durante la administración del tratamiento. En estos casos, podría ser útil la administración de terbinafina (38).
Hay que tener en cuenta que la malabsorción asociada al SPA tipo 1, puede complicar el tratamiento de la insuficiencia suprarrenal y del hipoparatiroidismo.
Está comenzando a plantearse la posibilidad de administrar tratamientos profilácticos antes de que aparezca las distintas patologías del síndrome pluriglandular: insulina en pacientes con riesgo de padecer diabetes mellitus y cortisol en los que pudieran desarrollar una insuficiencia suprarrenal primaria.
Bibliografía
1. Neufeld M, Maclaren NK, Blizzard RM. Two types of autoimmune Addison´s disease asssociated with different polyglandular autoimmune (PGA) syndromes. Medicine 1981; 60: 355-362. [ Links ]
2. Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev 2002; 23: 327-364. [ Links ]
3. Betterle C, Zanchetta R. Update on autoimmune polyendocrine syndromes (APS). Acta Biomed Ateneo Parmense 2003; 74: 9-33. [ Links ]
4. Bahceci M, Tuzcu A, Pasa S, Ayyildiz D, Tuzcu S. Polyglandular autoimmune syndrome type III accompanied by common variable immunodeficiency. Gynecol Endocrinol 2004; 1981: 47-50. [ Links ]
5. Hugle B, Dollmann R, Séller E, Kiess W. Addison's crisis in adolescent patients with previously diagnosed diabetes mellitus as manifestation of polyglandular autoimmune syndrome type II-report of two cases. J Pediatr Endocrinol Metab 2004; 17: 93-7 [ Links ]
6. Muir A, She JX. Advances in the genetics and immunology of autoimmune polyglandular syndrome II/III and their clinical aplications. Ann Med Interne 1999; 150: 301-312. [ Links ]
7. Whitaker J, Landing BH, Esselborn VM, Williams RR. The syndrome of familial juvenile hypoadrenocorticism, hypoparathyroidism and superficial moniliasis. J Endocrinol 1956; 16: 1374-1387. [ Links ]
8. Nagamine K, Peterson P, Scott H et al. Positional cloning of the APECED gene. Nat Genet 1997; 17: 393-8. [ Links ]
9. Aaltonen J, Bjorses P, Sandkuijl L, Perheentupa J, Peltonen L. An autosomal locus causing autoimmune disease. Autoimmune polyglandular disease type I assigned to chromosome 21. Nat Genet 1994; 8: 83-87. [ Links ]
10. Brun JM. Juvenile autoimmune polyendocrinopathy. Horm Res 1982; 16: 308-316. [ Links ]
11. Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Critical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med 1990; 322: 1829-1836. [ Links ]
12. Betterle C, Greggio NA Volpato M. Clinical review: autoimmune polyglandular disease type 1. J Clin Endocrinol Metab 1998; 83: 1049-1055. [ Links ]
13. Myhere AG, Halonen M, Eskelin P, Ekwall O, Hedstrand H, Rorsman F, Kämpe O, Husebye ES. Autoimmune polyendocrine syndrome type 1 (APS I) in Norway. Clin Endocrinol (Oxf) 2001; 54: 211-217. [ Links ]
14. Perheentupa J, Miettinen A. Type 1 autoimmune polyglandular disease. Ann Med Interne (Paris) 1999; 150: 313-325. [ Links ]
15. López-Jornet P, García-Ballesta C, Pérez-Lajarín L. Mucocutaneous candidiasis as first manifestation of autoimmune polyglandular syndrome type I. J Dent Child (Chic) 2005; 72: 21-4. [ Links ]
16. Bhansalli A, Kotwal N, Suresh V, Murlidharar R, Chattopadhyay A, Mathur K. Polyglandular autoimmune syndrome type 1 without chronic candidiasis in a 16 year-old-male. J Pediatr Endocrinol Metab 2003; 16: 103-5). [ Links ]
17. Dittmar M and Kahaly GJ. Polyglandular autoimmune syndromes: Immunogenetics and long-term follow-up. J Clin Endocrinol Metab 2003; 88: 2983-2992. [ Links ]
18. Lovas K, Husebye ES. High prevalence and increasing incidence of Addison´s disease underestimated? J Clin Endocrinol (Oxf) 2000; 56: 787-791. [ Links ]
19. Colls J, Betterle C, Volpato M, prentice L, Rees Smith B, Furmaniak J. A new immunoprecipitation assay for autoantibodies to steroid 21-hydroxylase in Addison's disease. Clin Chem 1995; 41: 375-380. [ Links ]
20. Chen S, Sawicka S, Betterle C et al. Autoantibodies to steroidogenic enzimes in autoimmune polyglandular syndrome. Addison´s disease and premature ovarian failure. J Clin Endocrinol Metab 1996; 81: 1871-1876. [ Links ]
21. Peterson P, Perheentupa J, Krhon KJE. Detection of candidal antigens in autoimmune polyglandular syndrome type I. Clin Diagn Lab Immunol 1996; 3: 290-294. [ Links ]
22. Report of a WHO Scientific Group. Primary immunodeficiencies diseases. Clin Exp Immunol 1995; 99 (Supl. 1): 1-24. [ Links ]
23. DePadova-Elder SM, Ditre CM, Cantor GR, Koblenzer PJ. Candidiasis endocrinopathy syndrome. Arch Dermatol 1994; 130: 19-22. [ Links ]
24. Li Y, Song Y, Rais N et al. Autoantibodies to the extracellular domain of the calcium sensing receptor in patients with acquired hypoparathyroidism. J Clin Invest 1996; 97: 910-914. [ Links ]
25. Betterle C, Volpato M. Adrenal and ovarian autoimmunity. Eur J Endocrinol 1998; 138: 16-25. [ Links ]
26. Muir A, MacLaren NK. Autoimmune diseases of the adrenal glands, parathyroid glands, gonads, and hypothalamic-pituitary axis. Endocrinol Metab Clin North Am 1991; 20: 619-644. [ Links ]
27. Tuomi T, Björses P, Falorni A et al. Antibodies to glutamic acid decarboxylase and insulin-dependent diabetes in patients with autoimmune polyendocrine síndrome type I. J Clin Endocrinol Metab 1996; 81: 1488-1494. [ Links ]
28. Betterle C, Volpato M, Greggio NA, Presotto F. Type 2 polyglandular autoimmune disease. J Pediatr Endocrinol Metab 1996; 9: 113-123. [ Links ]
29. Betterle C, Volpato M, Rees Smith B, Furmaniak J, Chen S, Zanchetta R, Greggio NA, Pedini B, Boscaro M, Presotto F. Adrenal cortex and steroid 21-hydroxylase autoantibodies in children with organ-specific autoimmune diseases: markers of high progression to clinical Addison´s disease. J Clin Endocrinol Metab 1997; 82: 939-942. [ Links ]
30. Ten S, New M, MacLaren N. Clinical review 130: Addison's disease. J Clin Endocrinol Metab 2001; 86: 2909-2922. [ Links ]
31. Jacobson DL, Gange SJ, Rose NR, Graham NMH. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol 1997; 84: 223-43. [ Links ]
32. Yamaguchi Y, Chikuba N, Ueda Y et al. Islet cell antibodies in patients with autoimmune thyroid disease. Diabetes 1991; 40: 319-22. [ Links ]
33. Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335: 1206-1212. [ Links ]
34. Schatz DA, Winter WE. Autoimmune polyglandular syndrome II. Clinical syndrome and treatment. Endocrinol Metab Clin N Am 2002; 31: 339-52. [ Links ]
35. Canaby A, Gieseler R, Ella R, Fink H, Saller B, Mann K. Manifestation of adrenal insufficiency after administration of levothyroxine in a patient with polyglandular autoimmune syndrome type II (Schmidt-syndrome) Internist (Berlin) 2000; 41: 588-591. [ Links ]
36. Graves L3rd, Klein RM, Walling AD. Addisonian crisis precipitated by thyroxine therapy: a complication of type 2 autoimmune polyglandular syndrome. South Med J 2003; 96: 824-7. [ Links ]
37. Kuriakose R, Koshy C. Anesthetic management of autoimmune polyglandular syndrome (Schmidt´s syndrome)-a case report. Middle East J Anesthesiol 2005; 18: 639-46. [ Links ]
38. Hassan G. Terbinafine effectiveness in ketoconazole-resistant mucocutaneous candidiasis in polyglandular autoimmune syndrome type 1. J Assoc Physicians India 2003; 51: 323. [ Links ]
39. FJ Candel González, M Matesanz David, I Candel Monserrate. Insuficiencia corticosuprarrenal primaria. Enfermedad de Addison. An Med Interna (Madrid) 2001; 18: 492-8). [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
María José Molina Garrido
Hospital General Universitario de Elche.
Camino de la Almazara, 11.
03203 Elche (Alicante).
mjmolinagarrido@hotmail.com
Trabajo aceptado: 18 de abril de 2007

