










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 n.1 Madrid Jan. 2004
|
RECOMENDACIONES DE PRÁCTICA CLÍNICA |
Abordaje diagnóstico y terapéutico del síndrome colestásico
T. Pérez Fernández, P. López Serrano, E. Tomás, Mª L. Gutiérrez, J. L. Lledó, G. Cacho, C. Santander
y C. M. Fernández Rodríguez
Unidad de Aparato Digestivo. Fundación Hospital Alcorcón. Madrid
RESUMEN
Ante la presencia de colestasis, se debe determinar si su naturaleza es extra o intrahepática. Si la ecografía hepática no muestra dilatación de la vía biliar ni lesiones ocupantes de espacio, se debe iniciar el estudio de una colestasis intrahepática. Si la obstrucción de la vía biliar extrahepática es cuestionable o la probabilidad de intervencionismo terapéutico es baja, se debe completar el estudio mediante colangio-pancreatografía-RM (CPRM). Si la probabilidad de intervencionismo es alta, se debe realizar colangiopancreatografía retrógrada endoscópica (CPRE) o colangiografía transparieto-hepática (CTPH). En caso de colestasis intrahepática, determinadas situaciones específicas ayudan a orientar el diagnóstico. Si la colestasis intrahepática ocurre en ancianos, se debe sospechar colestasis por fármacos, mientras que en pacientes jóvenes con antecedentes de riesgo, la hepatitis viral es la causa más frecuente. En el primer trimestre del embarazo la hiperemesis gravídica es la causa más probable y en el segundo o tercero la colestasis gravídica. La historia familiar y el curso recurrente deben orientar hacia una colestasis intrahepática recurrente benigna. La presencia de colestasis intrahepática en una mujer de edad media debe hacer sospechar CBP, mientras que en un varón joven con EIIC, una colangitis esclerosante primaria. La presencia de arañas vasculares, ascitis e historia de abuso de alcohol, apuntan hacia una hepatitis alcohólica como causa más probable. En el periodo neonatal, los síndromes colestásicos incluyen infecciones por CMV, toxoplasma, rubeola o defectos metabólicos como la fibrosis quística, el déficit de α1-antitripsina, defectos en la síntesis de ácidos biliares o atresia biliar. El tratamiento de la colestasis debe incluir el manejo de complicaciones como el prurito, la osteopenia y el déficit de vitaminas liposolubles. En caso de insuficiencia hepatocelular o complicaciones de la hipertensión portal, el manejo es similar al de otras etiologías y se debe valorar el trasplante hepático
Palabras clave: Coelstasis. Ultrasonografía. Colangio-pancreatografía. Prurito. Ácido ursodeoxicólico.
INTRODUCCIÓN
Colestasis es un defecto en la excreción biliar, en ocasiones acompañado de sintomas y signos clínicos como el prurito y la ictericia y trastornos bioquímicos como elevación de la fosfatasa alcalina (FA) (1).
Se distingue entre colestasis intra y extrahepática, según donde se localice la alteración al flujo biliar. En el primer caso la localización puede estar situada en cualquier punto entre el citoplasma de los hepatocitos y los conductos biliares de mediano calibre (100-400 µm). En las colestasis extrahepáticas la lesión obstructiva se encuentra en los conductos biliares grandes. En la mayoría de los casos es debido a litiasis o a tumores pancreato-biliares, incluyendo ampulomas o adenopatías hiliares (1).
CAUSAS DE COLESTASIS INTRAHEPÁTICA
1. Un grupo de estas causas se engloba bajo la denominación de síndromes de los conductos biliares evanescentes. Los más frecuentes son (Fig. 1):
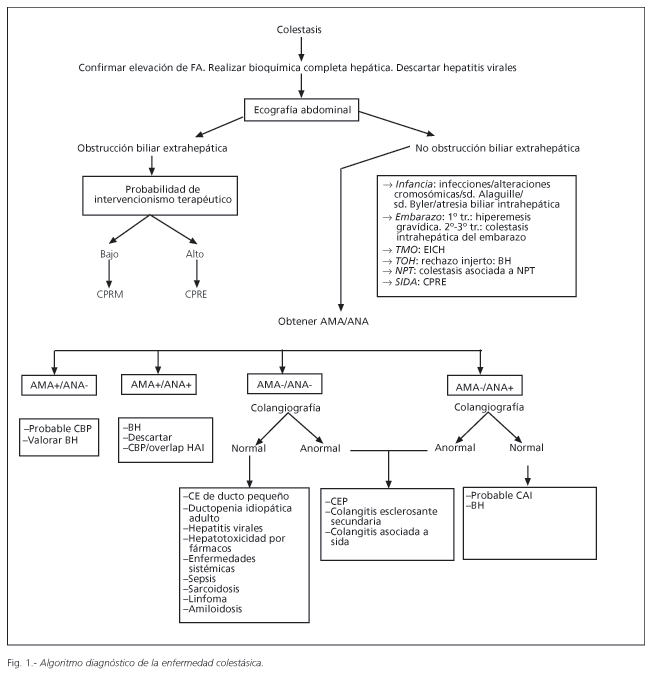
A. La cirrosis biliar primaria (CBP). La CBP se caracteriza por inflamación portal, necrosis del epitelio biliar y destrucción de los conductos biliares interlobulares y septales. El 90% de los pacientes son mujeres, de entre 40 y 65 años (2). En el 95% de los pacientes se detectan anticuerpos contra un antígeno de la membrana interna de la mitocondria (AMA), del complejo piruvato-deshidrogenasa (CPD), llamado E2. También se han encontrado asociados a la CBP, los autoantígenos gp210, p62, Sp100 y PML (3,4). Aunque la mayoría de los datos sugieren que la CBP es secundaria a una alteración de la inmunorregulación, su patogenia es incierta y no hay evidencia de que los anticuerpos antimitocondriales intervengan directamente en la lesión hepática. Se ha postulado una etiología infecciosa, ya que la proteína E2 está presente también en algunas bacterias y levaduras que colonizan a menudo las vías urinarias de las mujeres (3).
B. La colangitis esclerosante primaria (CEP) es también un síndrome colestásico crónico, de etiología desconocida y caracterizado por la obliteración fibrosa progresiva del árbol biliar, intra y extrahepático. Aparece típicamente en varones, siendo la edad media de 40 años (5). La mayoría de los pacientes tienen una enfermedad inflamatoria intestinal (6). Se postulan mecanismos patogénicos no inmunes (bacteriemia portal, virus, tóxicos o isquemia) y genéticos e inmunológicos. Entre el 26-85% de los pacientes presentan anticuerpos contra el citoplasma perinuclear de neutrófilos (p-ANCA) (4).
C. La colangitis autoinmune describe un grupo específico de pacientes en los que existen criterios clínicos y bioquímicos de colestasis e histología compatible con CBP, pero con negatividad de los anticuerpos AMA. Hasta en un 95% de los casos existen anticuerpos ANA y/o ASMA, y se han descrito anticuerpos contra un subtipo de la anhidrasa carbónica (CA-II), enzima presente en el epitelio del conducto biliar de los pacientes (7,8). Debe hacerse diagnóstico diferencial con los síndromes de solapamiento (CBP/ HAI y CEP/HAI).
D. La ductopenia idiopática del adulto afecta predominantemente a adultos jóvenes. Existen datos clínicos y bioquímicos de colestasis y el hallazgo en la biopsia hepática de desaparición de los conductos biliares, septales e interlobares, en más del 50% de los tractos portales (9,10).

Otros cuadros en los que existe también alteración en los conductos biliares intrahepáticos (11) son:
E. El rechazo del injerto tras trasplante hepático (12).
F. La enfermedad injerto contra huésped (EICH), donde la diana principal son los conductos biliares de pequeño tamaño (12),
G. Aproximadamente un 10% de pacientes con linfoma desarrollan ictericia, secundario a ductopenia y fibrosis periductal (13).
2. En muchas ocasiones la colestasis está relacionada con tóxicos hepáticos o trastornos metabólicos:
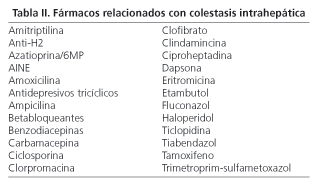
A. Colestasis inducida por fármacos y hormonas. Es frecuente en pacientes de edad avanzada. Los fármacos pueden producir daño hepático, habitualmente mediante un mecanismo inmunoalérgico. En ocasiones la lesión puede evolucionar hacia la ductopenia, remedando morfológicamente a la CBP (14,15). Aunque casi cualquier fármaco puede producir colestasis, algunos están implicados con mayor frecuencia (Tabla II).
B. Colestasis del embarazo. Existen dos tipos de trastornos durante el embarazo asociados a colestasis: la hiperemesis gravídica y la colestasis intrahepática del embarazo (16).
C. Nutrición parenteral total. El mecanismo por el que la NPT puede causar colestasis es múltiple: la administración de soluciones concentradas de glucosa y aminoácidos disminuyen el flujo biliar; el ayuno disminuye la excreción de ácidos biliares; el sobrecrecimiento bacteriano; y la posible coexistencia de infecciones y toxicidad por fármacos (11).
3. Las infecciones bacterianas pueden desencadenar también colestasis. La prevalencia de colestasis en pacientes con septicemia varía entre el 1 y el 34%. El trastorno puede deberse al efecto de toxinas bacterianas y citoquinas proinflamatorias, que pueden dar lugar a alteración en el transporte de la bilirrubina, disminución del recambio de ácidos biliares, aumento de la permeabilidad biliar y disminución del flujo biliar (1).
4. Los datos de colestasis son también comunes en las hepatitis.
A. En las hepatitis virales se asocian colestasis prolongadas por VHA, VHB y CMV. Hasta un 20% de los pacientes trasplantados por hepatitis crónica B desarrollan colestasis progresiva que evoluciona hacia el fallo hepático, denominado hepatitis colestásica fibrosante (17).
B. La colestasis puede ser un rasgo dominante tanto en la hepatitis autoinmune como en la hepatitis alcohólica.
5. Colestasis intrahepática benigna recurrente. Es un trastorno infrecuente caracterizado por episodios repetidos de colestasis, separados por periodos asintomáticos. Entre el 10-15% de los pacientes tienen antecedentes familiares y parece heredarse de forma recesiva (18).
6. Infiltración hepática. Algunas enfermedades pueden producir colestasis intrahepática por depósito en el parénquima hepático, o por infiltración tumoral. Un tercio de los pacientes con amiloidosis primaria tienen evidencia clínica o bioquímica de afectación hepática, secundaria a los depósitos de amiloide en las áreas periportales, aunque sólo un 5% de los pacientes presenta ictericia marcada (1).
SOSPECHA CLÍNICA DE COLESTASIS
Los síntomas y signos clínicos y alteraciones bioquímicas de la colestasis derivan de la acumulación de sustancias, habitualmente excretadas en la bilis, en el hígado, sangre y otros tejidos, y de la malabsorción de grasas y vitaminas liposolubles por disminución de ácidos biliares en el intestino delgado. Las colestasis intrahepáticas crónicas pueden manifestarse por la aparición de astenia, prurito, xantomas, diarrea, déficit de vitaminas liposolubles o fracturas vertebrales por osteoporosis (Tabla III). Con el paso de los años suelen evolucionar a cirrosis hepática e insuficiencia hepatocelular (11,19,20).

ACTITUD DIAGNÓSTICA ANTE UNA ENFERMEDAD COLESTÁSICA
La figura 1 representa una herramienta de aproximación al diagnóstico etiológico del síndrome colestásico. El primer paso es la elaboración de una correcta historia clínica, recogiendo todos los antecedentes personales y familiares de interés, tratamientos farmacológicos, uso de hierbas medicinales y abuso de alcohol, así como antecedentes epidemiológicos de riesgo para hepatitis virales y antecedentes familiares. Es importante confirmar la alteración en la analítica hepática antes de continuar el proceso diagnóstico (4,20) (Fig. 1).
El siguiente será la realización de una ecografía abdominal para la exclusión de patología biliar extrahepática. En el caso de detectar dilatación de la vía biliar, si la obstrucción de la vía biliar extrahepática es cuestionable o existe baja probabilidad de intervencionismo terapéutico, deberá realizarse una CPRM. Por el contrario, si las posibilidades de terapéutica biliar son altas, se debe completar el estudio mediante CPRE o CTPH. Cuando la ictericia es fluctuante y de baja intensidad (generalmente <12 mg/d), existe dolor abdominal y fiebre o antecedentes de cólicos biliares, el diagnóstico más probable es coledocolitiasis. Por el contrario, si la ictericia es indolora, progresiva e intensa (BT>12 mg/dl), existe alto riesgo de encontrar patología maligna.
Si no existen datos de imagen de patología biliar extrahepática, la orientación diagnóstica deberá establecerse en base a la historia clínica. Ante un paciente con situaciones específicas, como embarazo, trasplante hepático o de médula ósea o tratamiento con NPT, se deben sospechar las patologías más frecuentes asociadas a estas circunstancias (11). Si existe consumo de una medicación hepatotóxica, se recomienda la suspensión de la medicación y realizar un control posterior (11). La presencia de arañas vasculares, ascitis e historia de abuso de alcohol, apuntan hacia una hepatitis alcohólica como causa más probable. Los antecedentes epidemiológicos de riesgo para hepatitis virales orientan a este diagnóstico etiológico.
En caso de no existir antecedentes de interés ni datos de imagen de obstrucción biliar, las siguientes investigaciones deberán incluir una bioquímica completa con perfil férrico, cobre, alfa1-antitripsina, serología viral y screening de autoanticuerpos no órgano específicos [antinucleares (ANA), anti músculo liso (ASMA) y antimitocondriales (AMA)] (3).
La existencia de AMA positivos a un título mayor de 1/80 en una mujer de edad media con datos analíticos de colestasis apunta al diagnóstico de CBP. En la mayoría de los casos el diagnóstico se realiza tras el hallazgo casual de un aumento de la fosfatasa alcalina sérica y/o de anticuerpos antimitocondriales (21). El diagnóstico se basa en los siguientes criterios: a) fosfatasa alcalina sérica elevada al menos dos veces el valor normal; b) presencia de AMA positivos; y c) biopsia hepática con lesiones ductales biliares floridas (no necesaria si existen datos clínicos/bioquímicos de colestasis y positividad de los AMA en pacientes de mediana edad y sexo femenino) (22).
Si los AMA son negativos estará indicada la realización de una CPRM o CPRE para descartar la existencia de una CEP (4).
En el caso de colangiografía normal y ANA/ASMA positivos se debe considerar la biopsia hepática ante la sospecha de colangitis autoinmune o CBP AMA-negativa. Si la colangiografía es patológica, el primer diagnóstico a considerar es el de CEP. La presentación clínica de este cuadro puede variar desde una elevación asintomática de la fosfatasa alcalina sérica, hasta una ictericia colestásica, habitualmente en un varón joven con antecedentes de EIIC. El diagnóstico se basa en los hallazgos de la colangiografía, donde se observan múltiples estenosis de los conductos biliares alternando con segmentos normales, o dilataciones aneurismáticas. La biopsia hepática revela los cambios característicos de esta entidad, escasez de conductos biliares interlobulillares y fibrosis periductal en un tercio de los pacientes, por lo que es menos útil para el diagnóstico (22). Ante una colangiografía patológica, con cambios similares a la CEP, existen otras entidades menos frecuentes que deben ser descartadas clínicamente, como la colangitis esclerosante secundaria o la colangiopatía asociada a sida.
Si durante el estudio, tanto los autoanticuerpos como la colangiografía son negativos, los hallazgos de la biopsia hepática pueden orientarnos hacia una CEP de pequeño ducto o una ductopenia idiopática del adulto. El diagnóstico diferencial incluirá también otros trastornos como hepatitis virales, hepatotoxicidad por fármacos, colestasis intrahepática recurrente benigna o enfermedades sistémicas, como linfoma, sarcoidosis o amiloidosis. Existe una escala diagnóstica de lesiones hepáticas inducidas por fármacos drogas (23) de utilidad en el diagnóstico de las lesiones hepáticas de etiología tóxica.
MANEJO TERAPÉUTICO DE LA COLESTASIS
En el tratamiento de la colestasis crónica se debe valorar: a) el tratamiento de las complicaciones asociadas, que incluye la prevención y corrección del prurito, la enfermedad ósea metabólica, la malabsorción, el déficit de vitaminas liposolubles, la hipercolesterolemia y los xantomas; y b) el tratamiento específico de las enfermedades causales, incluyendo el trasplante hepático.
TRATAMIENTO DEL PRURITO
El desconocimiento de la patogenia del prurito ha obstaculizado el desarrollo de un tratamiento uniformemente efectivo. Aunque se ha propuesto que la acumulación cutánea de sales biliares puede ser la causa del prurito (24,25), su concentración no se correlaciona con la intensidad del prurito (26). También se ha sugerido que la concentración alta de sales biliares en el hígado provoca la rotura de la membrana celular y liberación de componentes pruritogénicos al torrente sanguíneo (27). Por otro lado, cada vez existe mayor evidencia del papel de los opioides endógenos en la patogenia del prurito de la colestasis (28). La serotonina también parece ser un mediador del prurito colestásico, probablemente modulando la acción opioide central (29).
El prurito leve puede responder al tratamiento con colestiramina de 4 a 16 gramos diarios v.o., esta resina de intercambio aniónico, secuestra las sales biliares intestinales e inespecíficamente sustancias pruritógenas que son eliminadas con las heces interrumpiendo su circulación enterohepática (24). Sus principales efectos secundarios son, estreñimiento, malabsorción de grasas y vitaminas liposolubles e interferencia con la absorción de digoxina, warfarina, propranolol, tiazidas y tiroxina; por lo que esta medicación debe ser administrada 1 hora antes o 4 horas después de la colestiramina.
La rifampicina es un enzimoinductor, aumenta la sulfoxidación de los ácidos biliares y por tanto su eliminación renal, aumenta el metabolismo de sustancias pruritogénicas, compite con los ácidos biliares por la captación hepática y minimiza la toxicidad de los ácidos biliares sobre el hepatocito. Dos estudios controlados han demostrado la mejoría del prurito de la colestasis con rifampicina a dosis de 300-600 mg/día en adultos (30,31) y un tercer estudio comparativo frente fenobarbital a dosis de 3 mg/kg de peso, que mostró mejoría significativa del prurito. Además, a diferencia del fenobarbital, la rifampicina mejoró la colestasis (32). Los efectos secundarios más frecuentes en tratamientos prolongados con rifampicina, ocurren en el 10% de los casos. Es el fármaco de elección en los pacientes que no responden a la colestiramina, si bien, es necesaria una monitorización de las pruebas de función hepática durante su uso.
El fenobarbital se ha utilizado a dosis única nocturna creciente hasta un máximo de 100 mg. Debido a su efecto sedante, no debe administrarse durante el día ni a pacientes con encefalopatía hepática. Los ensayos clínicos incluyen un número de pacientes pequeño y su eficacia es inferior a la rifampicina (32). Por lo que su uso no se puede recomendar en la actualidad.
El enzimoinductor flumecinol, a dosis de 600 mg/día durante tres semanas ha demostrado un ligero beneficio sobre el placebo en el tratamiento del prurito de la colestasis (33).
Los antagonistas opioides como la naloxona i.v. (0,2 µg/kg/minuto x 24 horas), el nalmefene oral (60-120 mg/día) y la naltrexona oral (50 mg/día) alivian el prurito de la colestasis (30-32). Estos resultados indican que la administración oral de nalmefene puede ser efectiva, si bien no existen ensayos clínicos controlados que confirmen esta posibilidad.
Aunque existen estudios no controlados con pequeño número de pacientes que indican la mejoría del prurito con fototerapia con luz ultravioleta (UV-B), plasmaféresis de gran volumen, el dispositivo MARS (molecular absortion and recirculating system) y andrógenos, como la metil-testosterona, la eficacia de estas modalidades terapéuticas es dudosa y no se recomiendan para el prurito que no responde a los fármacos convencionales.
Se han comunicado con un número muy reducido de casos, mejoría del prurito con ondansetrón, un antagonista serotoninérgico 5-HT3 (37,38). Recientemente, se ha demostrado un efecto superior en el alivio del prurito de la colestasis, aunque muy modesto frente a placebo (39).
El trasplante hepático está indicado en los casos de prurito incapacitante, que no responden a otras medidas, produce una rápida resolución del prurito y cura la enfermedad subyacente (17).
TRATAMIENTO DE LA MALABSORCIÓN
La colestasis intensa y prolongada puede originar malabsorción de grasas y de vitaminas liposolubles. Si aparece esteatorrea, se deben restringir las grasas de la dieta a 30 ó 40 g/día. En caso de desnutrición y pérdida de peso, se debe administrar suplementos con TCM, que no requieren sales biliares para su absorción.
TRATAMIENTO DE LA OSTEOPENIA Y DE VITAMINAS LIPOSOLUBLES
Las consecuencias clínicas del déficit de vitaminas liposolubles se recogen en la tabla III. En el caso de la vitamina A, se recomienda la administración de 50.000 UI cada 15 días. Es conveniente monitorizar periódicamente sus niveles para prevenir la hipervitaminosis A, que provoca astenia, letargia, molestias abdominales, anorexia, descamación cutánea, alopecia, hipertensión intracraneal y hepatotoxicidad. Se deben administrar a estos pacientes suplementos de calcio (1.500 mg al día) y vitamina D (266 µg de 25-hidroxi-colecalciferol cada una o dos semanas). Es necesaria una monitorización cuidadosa para comprobar la eficacia del tratamiento y para prevenir la sobredosificación de vitamina-D que produce hipercalcemia e hipercalciuria.
A diferencia de la población pediátrica, sólo una minoría de adultos con colestasis y niveles bajos de vitamina-E presentan síndrome neurológico de déficit de vitamina-E, que consiste en neuropatía periférica, degeneración cerebelosa, alteración en los movimientos oculares y retinopatía. Se recomienda administrar vitamina E: 100-200-400 UI/día ante la posibilidad de un beneficio aún no reconocido, sobre todo en pacientes con signos o síntomas neurológicos de etiología incierta.
Un tiempo de protrombina prolongado debido a la malabsorción de vitamina K se corrige con vitamina K s.c.10 mg/día, x 3 días. Después: suplementos crónicos via oral. 5-10 mg/día o subcutáneos 10 mg/mes.
TRATAMIENTO DE LA OSTEOPENIA
Debe realizarse una densitometría cuando se diagnostica una enfermedad colestásica crónica y posteriormente cada dos años. Para preservar la densidad ósea se recomienda la realización de ejercicio regular, exposición moderada a la luz solar, ingerir alimentos ricos en calcio y evitar el tabaco. Si la osteoporosis es evidente, se aconseja tratamiento con bifosfonatos. La administración cíclica de etidronato (400 mg/d durante dos semanas) en periodos de tres meses durante dos años, evita la pérdida de masa ósea en estas pacientes (41), aunque un estudio reciente no ha encontrado mejoría (42). Recientemente, el alendronato, a diferencia del etidronato, produjo un aumento de la densidad mineral ósea en un tratamiento durante 2 años (43).
La osteoporosis severa es indicación de trasplante hepático, incluso en ausencia de fallo hepático. Aunque la osteoporosis puede aumentar durante los primeros 6 meses post-trasplante, mejora considerablemente después (40). El tratamiento hormonal sustitutivo por vía transdérmica también puede prevenir la osteoporosis en las mujeres postmenopáusicas con colestasis (44).
ALTERACIÓN DEL METABOLISMO DE LOS LÍPIDOS
La hipercolesterolemia superior a 500 mg/dl, es frecuente en la CBP; sin embargo, la ateroesclerosis precoz es infrecuente. La dieta y la colestiramina son inefectivas para el tratamiento de la hipercolesterolemia. Dado el potencial hepatotóxico de muchos agentes hipolipemiantes y la falta de secuelas clínicas significativas de la hipercolesterolemia, debe considerarse su empleo en pacientes con colestasis que desarrollan complicaciones como xantomas dolorosos, pero no debe recomendarse de forma rutinaria. La plasmaféresis puede ser necesaria en pacientes con colesterol sérico >1.000 mg/dl.
TRATAMIENTO DEL FALLO HEPATOCELULAR
Los pacientes con enfermedades colestásicas avanzadas, en estadio cirrótico, pueden desarrollar síntomas y signos de fallo hepático, como ascitis, peritonitis bacteriana espontánea, encefalopatía hepática sangrado por varices esofágicas. El manejo es similar al empleado en otras etiologías de fallo hepático.
TRATAMIENTO DE LA ASTENIA
Algunos resultados preliminares sugieren que el tratamiento con ondansetrón (4 mg/8 h v.o.) disminuye la astenia en estos pacientes (45). El mecanismo podría estar relacionado con un efecto central en la neurotransmisión serotoninérgica.
TRATAMIENTOS ESPECÍFICOS DE LOS SÍNDROMES COLESTÁSICOS MÁS FRECUENTES
En el caso de la cirrosis biliar primaria (CBP) se han ensayado los esteroides, la azatioprina, la ciclosporina, el metotrexato, la D-penicilamina, la colchicina, el micofenolato y el bezafibrato, sin que el número de pacientes incluidos sea grande o el beneficio clínico observado sea consistente. La eficacia del ácido ursodeoxicólico (AUDC) se ha estudiado a dosis de 8-15 mg/kg/día por periodos de 3 meses a 5 años en 16 ensayos clínicos controlados, con un total de 1.422 pacientes. El resultado se ha resumido recientemente en un meta-análisis y una revisión sistemática del grupo Cochrane hepatobiliar (46,47). No se encontró efecto beneficioso en la mortalidad, OR: 0,94; IC 95% (0,60-1,48), ni en el trasplante hepático, OR 0,83; IC 95% (0,52-1,32) ni sobre ambos, OR: 0,9; IC 95% (0,65-1,26). Tampoco se encontró beneficio sobre el prurito, la astenia, la albúmina sérica ni la protrombina. Sin embargo, se redujo la ascitis, la ictericia, la bilirrubina sérica y las transaminasas. El análisis del subgrupo con enfermedad más avanzada o que recibió tratamiento más prolongado, no varió los resultados. Están en marcha estudios con dosis mayores de AUDC. Dado que este fármaco se asocia con pocos efectos secundarios y posiblemente ejerza un efecto beneficioso sobre la histología y algunos parámetros clínico-analíticos, es el tratamiento actual más utilizado (48).
El trasplante hepático está indicado en los pacientes con CBP con bilirrubina >6 mg/dl, signos de hipertensión portal (ascitis, encefalopatía, hemorragia digestiva) o que desarrollan hepatocarcinoma (40).
El tratamiento de la CEP incluye el tratamiento de las complicaciones locales, sépticas de origen biliar (mediante antibióticos ± tratamiento endoscópico) como de los cálculos biliares. Así como el tratamiento con AUDC (10-15 mg/kg/día) que mejora la bioquímica e histología de estos pacientes, aunque no está demostrado que obtengan beneficio clínico ni mejoría de la supervivencia (47). El trasplante hepático está indicado cuando aparecen episodios recurrentes de colangitis bacteriana, además de otras complicaciones como hiperbilirrubinemia persistente, complicaciones de la cirrosis y prurito refractario (40).
En la colangitis autoinmune, el AUDC produce mejoría bioquímica (aunque sin efecto en la supervivencia) (47). Algunos de estos pacientes con colangitis autoinmune, presentan solapamiento con la hepatitis autoinmune, y mejoran bioquímicamente con esteroides (solos o con azatioprina). No se han descrito marcadores que distingan los respondedores al AUDC o a la inmunosupresión, por lo que el tratamiento se basa en los hallazgos histológicos (49-53). En casos avanzados existe la opción del trasplante hepático.
En cuanto a la ductopenia idiopática del adulto, aunque el AUDC puede mejorar la bioquímica, no existen estudios suficientes para recomendarlo (54). En el resto de las colestasis hay que instaurar el tratamiento específico, como los inmunosupresores en la EICH y en el rechazo del trasplante hepático, la quimioterapia en la enfermedad de Hodgkin y la retirada del fármaco potencialmente hepatotóxico en las colestasis de origen farmacológico (55).
BIBLIOGRAFÍA
1. McIntyre N. Cholestasis. En: Bircher J, Benhamou J-P, McIntyre N, Rizzetto M, Rodés J, eds. Oxford textbook of Clinical Hepatology. 2th ed. Oxford: Oxford Medical. Publications, 1999. p. 1574-9. [ Links ]
2. Kaplan MM. Primary biliary cirrhosis. N Engl J Med 1996; 335: 1570-80. [ Links ]
3. Sherlock S, Heathcote J. Primary biliary cirrhosis. En: Bircher J, Benhamou J-P, McIntyre N, Rizzetto M, Rodés J, eds. Oxford textbook of Clinical Hepatology. 2th ed. Oxford: Oxford Medical. Publications, 1999. p. 1090-8. [ Links ]
4. Reshef R, Sbeit W, Lachter L. The chronic cholestasis enigma in adults. IMAJ 2002; 4: 449-53. [ Links ]
5. Ludwig J. Small-duct primary sclerosing cholangitis. Semin Liver Dis 1991; 11: 11-7. [ Links ]
6. Angulo P, Lindor KD. Primary sclerosing cholangitis. Hepatology 1999; 30: 325-32. [ Links ]
7. Lacerda MA, Ludwig J, Dickson ER, Jorgensen RA, Lindor KD. Antimitochondrial antibody-negative primary biliary cirrhosis. Am J Gastroenterol 1995; 90: 247-9. [ Links ]
8. Gordon SC, Quattrociocchi-Longe TM, Khan BA, Kodali VP, Chen J, Silverman AL, et al. Antibodies to carbonic anhydrase in patients with immune cholangiopathies. Gastroenterology 1995; 108: 1802-9. [ Links ]
9. Moreno A, Carreño V, Cano A, González C. Idiopathic biliary ductopenia in adults without symptoms of liver disease. N Engl J Med 1997; 336: 835-8. [ Links ]
10. Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc 1998; 73: 285-91. [ Links ]
11. Mc Gill JM, Kwiatkowski AP. Cholestatic liver diseases in adults. Am J Gastroenterol 1998; 93: 684-91. [ Links ]
12. Demetris AJ. Immune cholangitis: liver allograft rejection and graft-versus-host disease. Mayo Clin Proc 1998; 73: 367-79. [ Links ]
13. Perera DR, Greene ML, Fenster LF. Cholestasis associated with extrabiliary Hodgkin's disease. Report of three cases and review of four others. Gastroenterology 1974; 67: 680-5. [ Links ]
14. Ishak KG, Zimmerman HJ. Morphologic spectrum of drug-induced hepatic disease. Gastroenterol Clin North Am 1995; 24: 759-86. [ Links ]
15. De Leve LD, Kaplowitz N. Mechanisms of drug-induced liver disease. Gastroenterol Clin North Am 1995; 24: 787-810. [ Links ]
16. Lammert F, Marschall HU, Glantz A, Matern S. Intrahepatic cholestasis of pregnancy: molecular pathogenesis, diagnosis and management. J Hepatol 2000; 33: 1012-21. [ Links ]
17. Mason AL, Wick M, White HM, Benner KG, Lee RG, Regenstein F, et al. Increased hepatocyte expression of hepatitis B virus transcription in patients with features of fibrosing cholestatic hepatitis. Gastroenterology 1993; 105: 237-44. [ Links ]
18. Riely CA. Familial intrahepatic cholestatic syndromes. Semin Liver Dis 1987; 7: 119-33. [ Links ]
19. Kim WR, Ludwig J, Lindor KD. Variant forms of cholestatic diseases involving small bile ducts in adults. Am J Gastroenterol 2000; 95: 1130-8. [ Links ]
20. DGleeson D, Boyer JL. Intrahepatic cholestasis. En: Bircher J, Benhamou J-P, McIntyre N, Rizzetto M, Rodés J, eds. Oxford textbook of Clinical Hepatology. 2th ed. Oxford: Oxford Medical Publications, 1999. p. 1591-620. [ Links ]
21. Springer J, Cauch-Dudek K, O'Rourke K, Wanless IR, Heathcote EJ. Asymptomatic primary biliary cirrhosis: a study of its natural history and prognosis. Am J Gastroenterol 1999; 94: 47-53. [ Links ]
22. Poupon R, Chazouilleres O, Poupon RE. Chronic cholestatic diseases. J Hepatol 2000; 32 (Supl. 1): 129-40. [ Links ]
23. Aithal GP, Rawlins MD, Day CP. Clinical diagnostic scale: a useful tool in the evaluation of suspected hepatotoxic adverse drug reactions. J Hepatol 2000; 33: 949-52. [ Links ]
24. Datta DV, Sherlock S. Cholestyramine forlong term relief of the pruritus complicating intrahepatic cholestasis. Gastroenterology 1966; 50: 323-32. [ Links ]
25. Jschoenfield L, Sjovall J, Perman E. Bile acids on the skin of patients with pruritic hepatobiliary disease. Nature 1967; 213: 93-8. [ Links ]
26. Ghent CN, Bloomer JR, Klatskin G. Elevations in skin tissue levels of bile acids in human cholestasis: Relation to serum levels and to pruritus. Gastroenterology 1977; 73: 1125-30. [ Links ]
27. Ghent CN. Pruritus of cholestasis is related to effects to effects of bile salts on the liver, not the skin. Am J Gastroenterol 1987; 82: 117-8. [ Links ]
28. Jones EA, Bergasa NV. The pruritus of cholestasis and the opioid system. JAMA 1992; 268: 3359-62. [ Links ]
29. Jones E, Bergasa N. The pruritus of cholestasis. Hepatology 1999; 29: 1003-6. [ Links ]
30. Bergasa NV, Alling DW, Talbot TL, et al. Effects of naloxone infusions in patients with the pruritus of cholestasis. A double-blind, randomized, controlled trial. Ann Intern Med 1995; 123: 161-7. [ Links ]
31. Bergasa NV, Schmitt JM, Talbot TL, et al. Open-label trial of oral nalmefene therapy for the pruritus of cholestasis. Hepatology 1998; 27: 679-84. [ Links ]
32. Terg R, Coronel E, Sorda J, et al. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol 2002; 37: 717-22. [ Links ]
33. Ghent CN, Carruthers SG. Treatment of pruritus in primary biliary cirrhosis with rifampin. Results of a double-blind, crossover, randomized trial. Gastroenterology 1988; 94: 488-93. [ Links ]
34. Podesta A, López P, Terg R, Villamil F, Flores D, Mastai R, et al. Treatment of pruritus of primary biliary cirrhosis with rifampicin. Dig Dis Sci 1991; 36: 216-20. [ Links ]
35. Bachs L, Pares A, Elena M, Piera C, Rodes J. Comparison of rifampicin with phenobarbitone for treatment of pruritus in biliary cirrhosis. Lancet 1989; 1: 574-6. [ Links ]
36. Turner IB, Rawlins MD, Wood P, James OF. Flumecinol for the treatment of pruritus associated with primary biliary cirrhosis. Aliment Pharmacol Ther 1994; 8: 337-42. [ Links ]
37. Raderer M, Müller C, Scheithauer W. Ondansetron for pruritus due to cholestasis. N Engl J Med 1994; 330: 1540. [ Links ]
38. Schwörer H, Ramadori G. Improvement of cholestatic pruritus by ondansetron. Lancet 1993; 341: 1277 (Letter). [ Links ]
39. Muller C, Pongratz S, Pidlich J, Penner E, Kaider A, Schemper M, et al. Treatment of pruritus in chronic liver disease with the 5-hydroxytryptamine receptor type 3 antagonist ondansetron: a randomized, placebo-controlled, double-blind cross-over trial. Eur J Gastroenterol Hepatol 1998; 10: 865-70. [ Links ]
40. Neuberger J. Liver Transplantation for Cholestatic Liver Disease. Curr Treat Options Gastroenterol 2003; 6: 113-21. [ Links ]
41. Guañabens N, Pares A, Monegal A, Peris P, Pons F, Álvarez L, et al. Etidronate versus fluoride for treatment of osteopenia in primary biliary cirrhosis: preliminary results after 2 years. Gastroenterology 1997; 113: 219-24. [ Links ]
42. Lindor KD, Jorgensen RA, Tiegs RD, Khosla S, Dickson ER. Etidronate for osteoporosis in primary biliary cirrhosis: a randomized trial. J Hepatol 2000; 33: 878-82. [ Links ]
43. Pares A, Guañabens N, Ros I, Pons F, Álvarez L, Caballería L, et al. Alendronate versus etidronate for osteopenic patients with primary biliary cirrhosis: results after two years of treatment. J Hepatol 2001; 34 (Supl. 1): 19. [ Links ]
44. Le Gars L, Grandpierre C, Chazouilleres O, Berenbaum F, Poupon R. Bone loss in primary biliary cirrhosis: absence of association with severity of liver disease. Joint Bone Spine 2002; 69: 195-200. [ Links ]
45. Theal JJ, Toosi MN, Girlan LM, et al. Ondansetron ameliorates fatigue in patients with primary biliary cirrhosis (abstract). Hepatology 2002; 36: 296 A. [ Links ]
46. Goulis J, Leandro G, Burroughs AK. Randomized contrled trials of Ursodeoxycolic acid therapy for primary biliary cirrhosis. A meta-analysis. Lancet 1999; 354: 1053-60. [ Links ]
47. Gluud C, Christensen E. Ursodeoxycolic Acid for Primary Biliary Cirrhosis. (Cochrane review). Cochrane database Syst. Rev 2002; (1): CD000551. [ Links ]
48. Pares A, Caballería L, Rodes J, Bruguera M, Rodrigo L, García-Plaza A, et al. Long-term effects of ursodeoxycholic acid in primary biliary cirrhosis: results of a double-blind controlled multicentric trial. UDCA-Cooperative Group from the Spanish Association for the Study of the Liver. J Hepatol 2000; 32: 561-6. [ Links ]
49. Kim WR, Ludwig J, Lindor KD. Variant forms of cholestasis diseases involving small bile ducts in adults. Am J of Gastroenterol 2000; 95: 1130-8. [ Links ]
50. Ben Ari Z, Dhilon AP, Sherlock S. Autoinmune cholangiopathy: parts of the spectrum of autoinmmune hepatitis. Hepatology 1993; 18: 10-5. [ Links ]
51. Gisbert JP, Jones EA, Pajares JM, Moreno-Otero R. Review article: is there an optimal therapeutic regimen for antimitochondrial antibody-negative primary biliary cirrhosis (autoinmmune cholangitis). Aliment Pharmacol Ther 2003; 17: 17-27. [ Links ]
52. Reshef R, Sbit W, Laxhter J. The chronic cholestasis enigma in adults. IMAJ 2002; 4: 449-53. [ Links ]
53. Sánchez-Pobre P, González C, Paz E, Colina F, Castellano G, Muñoz-Yagüe T, et al. Chronic hepatitis C and autoinmmune cholangitis: a case study and litteratue review. Dig Dis Sci 2002; 47 (6): 1224-9. [ Links ]
54. Linares Rodríguez A. Tratamiento médico de la colestasis crónica: ácido ursodesoxicólico. Gastroenterol Hepatol 2000; 23 (Supl. 1): 24-38. [ Links ]
55. Qureshi WA. Intrahepatic cholestatic syndromes: pathogenesis, clinical features and management. Dig Dis 1999; 17: 49-59. [ Links ]

