










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.97 n.12 Madrid Dec. 2005
| NOTA CLÍNICA |
Enfermedad celiaca (EC), colitis ulcerosa (CU) y colangitis esclerosante primaria
(CEP) asociadas en el mismo paciente: estudio familiar
V. Cadahía, L. Rodrigo, D. Fuentes, S. Riestra, R. de Francisco y M. Fernández1
Servicios de Aparato Digestivo y de 1Anatomía Patológica. Hospital Universitario Central de Asturias. Oviedo
RESUMEN
Presentamos el caso de un varón de 17 años, que a la edad de 7 años fue diagnosticado de enfermedad celiaca (EC) junto con una colitis ulcerosa (CU) y una colangitis esclerosante primaria (CEP) asociadas. Fue tratado con DSG e inmuno-supresores tipo azatioprina y se encuentra asintomático en la actualidad.
Su hermana menor de 12 años, fue diagnosticada de EC cuando tenía 1,5 años y a los 7 años desarrolló una DM tipo 1 de difícil control.
Se realizó un estudio familiar y ambos padres están afectos de una EC silente. Todos ellos son DQ2 (+). A propósito del caso y estudio familiar, se hacen una serie de consideraciones sobre la enfermedad celiaca y el desarrollo de complicaciones.
Palabras clave: Enfermedad celiaca (EC). Colitis ulcerosa (CU). Colangitis esclerosante primaria (CEP). Estudio familiar.
INTRODUCCIÓN
La enfermedad celiaca (EC) es un proceso que se presenta en individuos genéticamente predispuestos, generalmente HLA-DQ2 (+) en alrededor del 90% de los casos, o HLA-DQ8 (+) en el 7-10% restante, como consecuencia de una respuesta inmune alterada frente a un factor extrínseco que es la gliadina, proteína contenida en la harina de la mayor parte de los cereales, con la única excepción del maíz.
En pacientes afectos con la EC, es relativamente frecuente encontrar la presencia de enfermedades asociadas como diabetes tipo I, dermatitis herpetiforme y tiroiditis autoinmune entre otras (1-3).
Igualmente es común el hallazgo de una moderada agregación familiar en pacientes con EC, que oscila entre el 10-20% de los casos (4), pudiendo encontrar familiares asintomáticos o con formas atípicas, en los que si no se realiza una búsqueda intencionada de casos, pueden permanecer sin diagnosticar, durante largos periodos de tiempo.
La colangitis esclerosante primaria (CEP) es una enfermedad hepática de naturaleza autoinmune, en la que están implicados factores genéticos, adquiridos, o ambos. Su asociación con otras enfermedades está muy demostrada y entre ellas la más frecuente es la enfermedad inflamatoria intestinal (EII), principalmente la colitis ulcerosa (CU) (en el 70% de los casos).
Presentamos el caso de un paciente con EC asociada con CU y CEP, comenzando todas ellas en la infancia y en cuyo estudio familiar encontramos diferentes formas de presentación de la EC junto con otras enfermedades asociadas en el padre y una hermana también afectos.
CASO CLÍNICO
Paciente de 17 años que acudió a nuestro centro por primera vez hace diez años, cuando tenía 7 años de edad, procedente de otro hospital comarcal, por presentar una alteración persistente de las pruebas de función hepática, de un año de evolución.
Como antecedentes de interés, referían sus padres la existencia de múltiples episodios diarreicos intermitentes, desde los seis meses de edad, que habían sido etiquetados como compatibles con un síndrome de colon irritable.
No refería antecedentes previos de transfusiones sanguíneas, episodios de hepatitis aguda, ictericia, ni prurito. La exploración física fue negativa, presentando el paciente un desarrollo estato-ponderal normal para su edad.
El hemograma fue normal y las PFH mostraban un patrón mixto de citolisis + colestasis disociada, con una AST = 133 (n < 40) UI/L; ALT= 211 (n < 40) UI/L; FA = 1.365 (n < 180) UI/L; GGT= 268 (n < 50) UI/L; Bil. total = 2,2 (n < 1) mg/dl.
La determinación de los diversos marcadores virales de hepatitis (frente al VA, VB, VC, CMV, EB, HS y VZ) así como los anticuerpos anti-toxoplasma fueron todos negativos, así como la determinación de auto-anticuerpos no-órgano específicos (ANAs, AMAs, AML y anti-LKM1) y el despistaje de diversas metabolopatías (hemocromatosis, enfermedad de Wilson y déficit de a1-antitripsina) fue igualmente negativo.
Los estudios analíticos mostraron además de la referida alteración de las PFH, la presencia de ANAs (+) a título bajo (1/40); EMAs (+) (1/80); anticuerpos anti-reticulina (+) (1/160) y p-ANCA (+) (1/320), todos ellos a títulos medios-altos, siendo el resto de las determinaciones analíticas normales.
Para el estudio de la diarrea y por la presencia de marcadores serológicos positivos de enfermedad celiaca, se le realizó una biopsia intestinal con cápsula de Watson-Crosby. El estudio histológico mostró la presencia de una marcada atrofia de las vellosidades duodenales, con hipertrofia de las criptas y porción basal, junto con marcado infiltrado linfocitario en la submucosa, confirmando el diagnóstico de EC y siendo clasificado como estadio 3c de Marsh (Fig. 1A).
Se le realizó una rectosigmoidoscopia que mostró la presencia de una mucosa edematosa, friable, fácilmente sangrante al contacto con el endoscopio, con múltiples erosiones y ulceraciones superficiales. Las biopsias de colon mostraron la presencia de un infiltrado inflamatorio crónico de la submucosa, con presencia de abscesos crípticos y erosiones epiteliales, siendo diagnosticado de colitis ulcerosa de grado moderado (Fig. 1B).
Se le practicó una biopsia hepática, en la que se objetivó la presencia de un infiltrado inflamatorio moderado portal y lobulillar, con intensa fibrosis porto-portal y periductular, en forma de capas de cebolla, junto con una proliferación ductulillar intensa, siendo informada como colangitis esclerosante primaria (CEP) (Fig. 1C).
Para realizar el diagnóstico topográfico y la extensión de esta última entidad, se le realizó una CPRE, que mostró la presencia de un árbol biliar intrahepático con pobreza en ramas canaliculares y con "ductos arrosariados", junto con una vía biliar extrahepática conservada, sin alteraciones llamativas, confirmando el predominio de la afectación intrahepática. (figura 2)
Se puso al paciente con dieta sin gluten (DSG) y se inició tratamiento con ácido ursodesoxicólico (900 mg/día) y 5-ASA (2 g/día), permaneciendo asintomático y con mejoría de las pruebas hepáticas durante 2 años. Pasado este tiempo, presentó un episodio de colangitis aguda, que evolucionó favorablemente con tratamiento antibiótico domiciliario y un brote de colitis ulcerosa que precisó ingreso hospitalario, a raíz del cual, se inició tratamiento con azatioprina (150 mg/día), que mantiene hasta la actualidad. Hace 2 años se le realizó una gastroscopia que fue normal, con toma de biopsias duodenales que fueron también completamente normales, sin signos de atrofia, ni de inflamación.
El paciente se encuentra en la actualidad completamente asintomático, realizando una vida completamente normal, siguiendo controles semestrales por nuestro servicio.
ESTUDIO FAMILIAR
Un año después de ser diagnosticado el caso índice, su hermana menor, a los 18 meses de edad, comenzó a presentar diarrea, dolor abdominal y vómitos, junto con adelgazamiento y retraso de crecimiento, por lo se realizaron estudios serológicos y biopsias yeyunales, con los que se confirmó el diagnóstico de enfermedad celiaca (EC), en su forma clásica, con biopsia duodenal con atrofia moderada (estadio 3b de Marsh). Se inició dieta sin gluten con buena respuesta clínico-analítica. Seis años más tarde, a la edad de 7,5 años, esta paciente presentó una diabetes mellitus tipo I, de difícil control metabólico, con necesidad de tres dosis diarias de insulina hasta la actualidad.
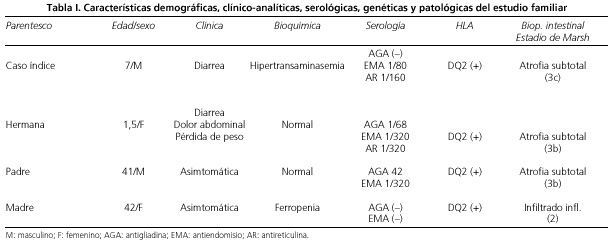
Con los hallazgos de ambos hermanos se procedió a realizar el estudio de EC a sus padres que estaban completamente asintomáticos, presentando analíticamente la madre sólo una discreta ferrropenia y el padre mostraba una analítica normal, con anticuerpos positivos y atrofia vellositaria moderada (estadio 3b de Marsh), siendo diagnosticados ambos de EC silente y puestos a DSG continuando estando ambos asintomáticos, sin enfermedades asocidas, ni mostrando alteraciones analíticas en la actualidad. Los resultados del estudio analítico, genético e histológico familiar muestran que todos los miembros de la familia están afectos, siendo los cuatro DQ2 (+), y la madre presenta una serología negativa y en la biopsia duodenal no muestra signos de atrofia vellositaria, sino únicamente infiltrado inflamatorio a nivel de la submucosa (estadio 2 Marsh) (Tabla I).
DISCUSIÓN
La asociación entre CU y CEP está bien documentada y se calcula que entre un 3-5% de pacientes con CU, pueden desarrollar una CEP a lo largo de su vida (4).
Sin embargo la asociación entre EC y CEP es mucho menor, estimándose una frecuencia media en torno al 1,5% (5).
Se ha descrito también una asociación entre EC y hepatopatías colestásicas crónicas autoinmunes, tipo cirrosis biliar primaria (CBP) y colangitis autoinmune (CAI) que se presentan con una frecuencia intermedia entre las anteriores, y está situada en torno al 3,5% de los casos (6).
Hasta la actualidad existen pocos casos descritos de esta rara asociación EC, CU y CEP. En la mayor parte de los casos, la colitis ulcerosa precede varios años al diagnóstico de las otras dos enfermedades, a diferencia de nuestro caso, en el que se diagnosticaron los tres procesos, prácticamente de forma simultánea.
Por ello, ante todo paciente con enfermedad celiaca, recomendamos realizar una búsqueda activa de enfermedades asociadas, no sólo digestivas, sino también sistémicas. Ello parece ser debido a que diversos procesos comparten un mismo haplotipo HLA de clase II condicionado genéticamente, lo que sería probablemente el principal factor favorecedor para el desarrollo de otras enfermedades concomitantes de naturaleza autoinmune (7,8) Se ha especulado acerca de si el retraso en efectuar el diagnóstico de la EC, y por tanto en la instauración de una dieta sin gluten (DSG), podría favorecer la aparición de enfermedades asociadas, aunque esta hipótesis está muy controvertida y por el momento no aclarada (9,10).
En un estudio llevado a cabo con el objetivo de estimar la prevalencia de enfermedad celiaca en niños y jóvenes con diabetes mellitus tipo 1 y la influencia de su diagnóstico precoz, para la prevención de un desarrollo posterior de EC, señalan que la prevalencia de esta asociación es del 7% que el diagnóstico de la DM tipo 1 a una edad precoz (< 4 años) y el sexo femenino, están asociados con un mayor riesgo de presentar ambas enfermedades. Existen también varios estudios con resultados contradictorios en cuanto a que el diagnóstico precoz de la EC y el inicio por tanto temprano de una dieta exenta de gluten, evite el desarrollo de otras enfermedades de base inmunológica (11,12).
En nuestra familia, la hija menor fue diagnosticada al año y medio de edad de enfermedad celiaca y pese a llevar una estricta DSG, desarrolló al cabo de cinco años de tratamiento una DM tipo 1. Todos los miembros de la familia eran HLA-DQ2 (+), que como es bien conocido es el gen que está íntimamente relacionado con el desarrollo de la enfermedad celiaca pero también con el de la diabetes mellitus tipo 1 (13).
Existe un riesgo aumentado del desarrollo de neoplasias malignas tipo colangiocarcinoma en los pacientes con CEP y de cáncer de colon en pacientes con pancolitis ulcerosa de comienzo juvenil, agudo grave, a partir de los 10 años de evolución, aunque hay trabajos recientes que señalan una posible prevención de este último, en pacientes tratados con ácido ursodesoxicólico a largo plazo (14).
Aunque la agregación familiar de la EC está entre un 10-20%, en nuestro caso resultaron afectos todos sus miembros, dos de ellos con un diagnóstico en la infancia y con enfermedades asociadas, mientras que los padres fueron diagnosticados de formas silentes de EC a través del estudio familiar y presentaron también una buena respuesta a la DSG (15).
Es muy importante por tanto llevar a cabo de forma rutinaria un estudio familiar ante todo diagnóstico de EC para el despistaje de nuevos casos, aunque se encuentren asintomáticos, ya que la instauración de una DSG puede evitar el desarrollo de complicaciones y prevenir la aparición de enfermedades asociadas.
BIBLIOGRAFÍA
1. Collin P, Kaukinen K, Välimäki M, Salmi J. Endocrinological disorders in celiac disease. Endocrine Reviews 2002; 23: 464-83. [ Links ]
2. Zone JJ. Skin manifestations of celiac disease. Gastroenterology 2005; 128 (Supl. 1): S87-91. [ Links ]
3. Hakanen M, Luotola K, Salmi J, et al. Clinical and subclinical autoimmune thyroid disease in adult celiac disease. Dig Dis Sci 2001; 46: 2631-5. [ Links ]
4. Mac Faul GR, Chapman RW. Sclerosing cholangitis. Curr Op Gastroenterol 2004; 20: 275-80. [ Links ]
5. Smyth C, Kelleher D, Keeling PW. Hepatic manifestations of gastrointestinal diseases. Inflammatory bowel disease, celiac disease and Whipple's disease. Clin Liver Dis 2002; 6: 1013-32. [ Links ]
6. Volta U, Rodrigo L, Granito A, et al. Celiac disease in autoimmune cholestatic liver disorders. Am J Gastroenterol 2002; 97: 2609-13. [ Links ]
7. Harbior A, Rawa T, Orlowska J, et al. Association of primary sclerosing cholangitis, ulcerative colitis and coeliac disease in female siblings. Eur J Gastroenterol Hepatol 2002; 14: 787-91. [ Links ]
8. Wurm P, Dixon AD, Rathbone BJ. Ulcerative colitis, primary cholangitis and coeliac disease: two cases and review of the literature. Eur J Gastroenterol Hepatol 2003; 15: 815-7. [ Links ]
9. Ventura A, Magazzu G, Greco L. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. Gastroenterology 1999; 117: 297-303. [ Links ]
10. Sategna C, Solerio E, Scaglione N, et al. Duration of gluten exposure in adult celiac disease does not correlate with the risk for autoimmune disorders. Gut 2002; 49: 502-5. [ Links ]
11. Cerruti F, Bruno G, Chiarelli F, et al. Younger age at onset and sex predict celiac disease in children and adolescents with type 1 diabetes. Diabetes Care 2004; 27: 1294-8. [ Links ]
12. Biagi F, Pezzimenti D, Campanella J, et al. Gluten exposure and risk of autoimmune disorders. Gut 2002; 50: 140-1. [ Links ]
13. Doolan A, Donaghue K, Fairchild J, Wong M, Williams AJ. Use of HLA typing in diagnosing celiac disease in patients with type 1 diabetes. Diabetes Care 2005; 28: 806-9. [ Links ]
14. Pardi DS, Loftus EV Jr, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 2003; 124: 889-93. [ Links ]
15. Rodrigo L, Riestra S, Fuentes D, González S, López-Vázquez A, López-Larrea C. Diversas formas clínicas de presentación de la enfermedad celíaca dentro de la misma familia. Rev Esp Enferm Dig 2004; 96: 612-9. [ Links ]

