










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.106 n.8 Madrid Dec. 2014
Síndrome LPAC secundario a deleción del exón 4 del gen ABCB4 en un paciente con hepatitis C
LPAC syndrome associated with deletion of the full exon 4 in a ABCB4 genetic mutation in a patient with hepatitis C
Blanca Fombuena1, Javier Ampuero1, Luis Álvarez2, Reyes Aparcero1, Rocío Llorca1, Raquel Millán1, Helena Pastor-Ramírez1, Sara Andueza2, Veronique Barbu3 y Manuel Romero-Gómez1
1UGMQ Enfermedades Digestivas y CIBERehd. Universidad de Sevilla. Hospital Universitario de Valme. Sevilla
2Unidad de Investigación. Hospital Universitario La Paz-IdiPAZ. Madrid
3Service de Biochimie, Biologie Cellulaire et Moléculaire. Hôspital Universitaire Saint-Antoine. París
RESUMEN
El síndrome LPAC (low-phospholipid-associated cholelithiasis syndrome) está asociado a mutaciones del gen ABCB4, que codifica la proteína MDR3, esencial en la secreción de fosfatidilcolina en las sales biliares. Este síndrome se caracteriza por una mayor prevalencia en mujeres, síntomas biliares en adultos jóvenes y excelente respuesta al ácido ursodesoxicólico (AUDC). Presentamos el caso de un hombre de 48 años con hepatitis C, genotipo 1b, fibrosis F3, nula respuesta Peg-IFN-α-2b/ribavirina y cólicos nefríticos de repetición. En 2011 desarrolló ictericia, prurito y dolor cólico epigástrico acompañado de aumento sérico de AST, ALT, GGT, bilirrubina y alfafetoproteína, y carga viral (14.600.000 UI/ml). La endoscopia oral, la ecoendoscopia, la angio-TAC y la ecografía-doppler evidenciaron hepatopatía crónica no cirrótica. El cuadro se autolimitó y un año después sufrió un episodio similar. Iniciamos tratamiento con AUDC, con excelente respuesta clínica. El estudio inmunohistoquímico y la secuenciación completa del gen ABCB4 no mostraron alteraciones. La técnica MLPA® detectó deleción heterocigota del exón 4 completo y confirmó la sospecha de síndrome LPAC.
Palabras clave: LPAC. ABCB4. MDR3. Exón 4.
ABSTRACT
Low-phospholipid-associated cholelithiasis syndrome (LPAC) is associated with ABCB4 genetic mutation. ABCB4 encodes MDR3 protein, involved in biliary phosphatidylcholine excretion. Higher prevalence in women, biliary symptoms in young adults and ursodesoxycholic acid (UDCA) response are the main features. We report the case of a 48-year-old man with hepatitis C, genotype 1b, fibrosis F3, null responder to Peg-IFNα2b/ribavirin and nephritic colic. In 2011 he developed jaundice, pruritus and epigastric pain. He showed increased serum levels of AST, ALT, GGT, bilirubin and alpha-fetoprotein, and viral load (14,600,000IU/mL). Pancreatic-CT, endoscopic ultrasonography and echo-Doppler showed non-cirrhotic chronic liver disease. The episode resolved spontaneously and one year later he suffered a similar episode. UDCA was started with excellent response. An immunohistochemistry study and sequencing of ABCB4 did not find alteration. MLPA® technique detected heterozygous deletion of the full exon 4 confirming LPAC syndrome diagnosis.
Key words: LPAC. ABCB4. MDR3. Exon 4.
Introducción
El síndrome LPAC (de sus siglas en inglés, low phospholipid-associated cholelithiasis) fue descrito por primera vez en 2001 como una forma peculiar de colelitiasis intrahepática ocurrida en adultos jóvenes, menores de 40 años, asociada a mutaciones del gen ABCB4 -localizado en el cromosoma 7q-21 (1)-, responsable de codificar MDR3 (2), perteneciente a la familia de las proteínas MDR (multidrug resistance protein). Se trata de una proteína transmembrana que actúa como una bomba dependiente de ATP expulsando una alta gama de moléculas pequeñas, siendo esencial para la secreción de fosfatidilcolina (FC) en la bilis (3). La FC, junto con el colesterol y los ácidos biliares, forman las micelas biliares que inactivan la acción detergente de las sales biliares y previenen el daño a las células epiteliales que recubren el conducto biliar, así como también inhiben la formación de cálculos biliares de colesterol (4). Una modificación en el gen ABCB4 conlleva una producción de bilis con un bajo contenido en fosfolípidos, una mayor producción de cálculos y elevadas propiedades detergentes, lo que causa lesiones en la membrana del lumen de los conductos biliares (5). Estas alteraciones genéticas están asociadas con la colestasis familiar progresiva intrahepática (PFIC3, del inglés progressive familiar intrahepatic cholestasis type 3), la colelitiasis asociada a un nivel bajo de fosfolípidos (LPAC) (6) y la colestasis intrahepática del embarazo (ICP del inglés intrahepatic cholestasis of pregnancy). Esta variedad de fenotipos se debe a que la mutación reduce pero no elimina la proteína permitiendo así una actividad residual del transportador. La colelitiasis es poco común en Europa, siendo más frecuente en Asia (7). Sobre un 10 % de la población europea y americana sufre cálculos biliares, aproximadamente un 25 % de los casos presenta síntomas y menos del 2 % presenta complicaciones severas (colangitis o pancreatitis). Desde la descripción de las mutaciones de MDR3 ha aumentado el número de pacientes diagnosticados con LPAC (8).
Caso clínico
Presentamos el caso de un varón de 48 años con antecedentes familiares de madre fallecida por hepatopatía crónica inespecífica evolucionada hacia cirrosis hepática y hermana con hepatopatía no filiada. En sus antecedentes personales destaca la presencia de hepatitis C (VHC) genotipo 1b con nula respuesta a peginterferón α-2a y ribavirina en dos ocasiones. Además, cólicos nefríticos de repetición.
En febrero de 2011 presentó un episodio de ictericia intensa, con prurito y epigastralgia asociada. Los valores analíticos más destacables fueron: AST 145 U/L (0-37 U/L), ALT 121 U/L (0-37 U/L), fosfatasa alcalina 116 U/L (50-190 U/L), GGT 167 U/L (6-50 U/L), bilirrubina total 14,6 mg/dl (0-1,8 mg/dl), y alfa-feto proteína 19,14 ng/ml (0-5 ng/ml), así como carga viral VHC de 14.600.000 UI ml. Se realizaron las siguientes pruebas diagnósticas: ecografía digestiva, en la que no se apreciaban lesiones ocupantes del espacio ni dilatación de las vías biliares y mostraba una bilis espesa sin datos concluyentes de colelitiasis; colangio-RM en la que se descartó coledocolitiasis y litiasis intrahepática; endoscopia oral normal; angio-TAC que mostró hepatomegalia homogénea y ecoendoscopia que descartó lesiones en el páncreas o la vía biliar. La biopsia hepática reveló una fibrosis grado 3 según METAVIR compatible con hepatitis C, sin otras alteraciones valorables.
Un año después (febrero 2012) vuelve a consulta presentando un episodio de similares características (ictericia, prurito y epigastralgia); ante la recurrencia del cuadro se prescribió AUDC, con una mejoría rápida del cuadro (Fig. 1).
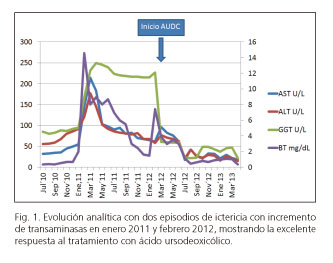
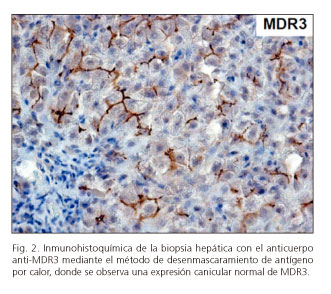
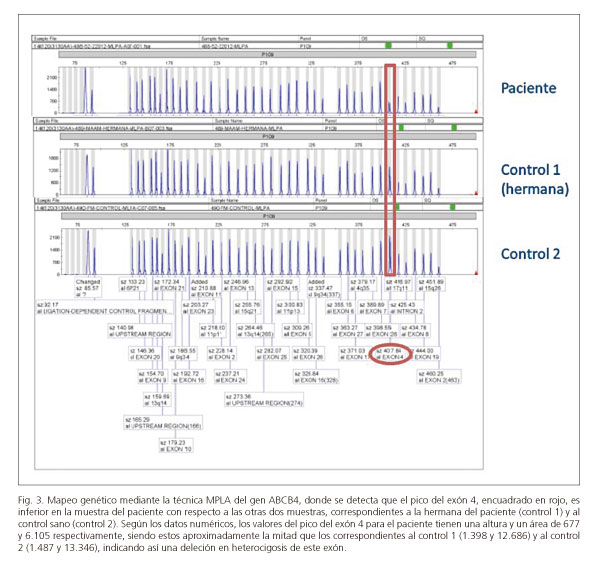
Ante la clínica recidivante y la ausencia de causa etiológica, se sospechó de un síndrome LPAC. Se envió una muestra al Hospital Universitario La Paz (Madrid) para estudio inmunohistoquímico de la biopsia hepática, apreciándose una expresión canicular normal de MRD3 (Fig. 2). Posteriormente se estudió la secuencia codificante del gen ABCB4 (muestra de ADN de linfocitos de sangre periférica) en el Hospital Universitario Saint-Antoine (París) mediante la tecnología Roche 454 GS Junior sequencing, siendo esta una nueva generación de secuenciación del ADN por medio de una secuenciación ultraprofunda de productos PCR, no encontrándose tampoco ninguna mutación. Ante la persistencia de la sospecha clínica, en el Hospital Universitario La Paz se analizó nuevamente el gen ABCB4 esta vez con la técnica MLPA® (Multiplex Ligation-dependent Probe Amplification) (9), tratándose de una técnica complementaria que detecta cambios en el número de copias genómicas, inserciones o deleciones no detectados por los métodos convencionales de la secuenciación. Se comparó la muestra del paciente con una muestra de su hermana (control 1) y la de un individuo sano (control 2). Se identificó una mutación en heterocigosis por la deleción de un fragmento de gen que contiene el exón 4 completo (Fig. 3), compatible con una deficiencia parcial de MDR3 (10), confirmándose el diagnóstico de LPAC.
Discusión
El síndrome LPAC es una rara entidad caracterizada por cálculos de colesterol intrahepáticos y colelitiasis, con repercusión clínica en forma de síntomas biliares, incluso a pesar de colecistectomía. LPAC se ha asociado a mutaciones en el gen ABCB4, que codifica la proteína MRD3. Sin embargo, estas mutaciones se observan en aproximadamente un 60 % de los casos (en un subgrupo de pacientes) (11), por lo que el diagnóstico se basa principalmente en criterios clínicos y ecográficos. Aunque se debería tener en cuenta que este porcentaje de pacientes, con mutaciones en ABCB4, sólo se refieren a las alteraciones en la secuenciación no a deleciones, por lo que el porcentaje podría ser mayor. En general, deberían existir al menos dos de los siguientes criterios: a) edad ≤ 40 años; b) reaparición de síntomas biliares tras colecistectomía; y c) focos hiperecogénicos intrahepáticos, barro biliar o microlitiasis (12). Otras características clínicas asociadas son: a) colestasis intrahepática en el embarazo; b) colelitiasis en parientes de primer grado; c) predominio en mujeres; y d) mejoría franca con AUDC, generalmente en seis meses. Esto último sugiere que los síntomas no están siempre directamente relacionados con la litiasis, sino con una inflamación subyacente de los conductos biliares intrahepáticos. Nuestro paciente se trata del primer caso reportado en España y, además, tiene algunas características singulares. En primer lugar, la mutación observada (deleción del exón 4) no ha sido previamente comunicada (13). Segundo, se trató de un varón mayor de 40 años, lo que supone una presentación atípica en cuanto a género y edad. Tercero, al tratarse de un paciente con hepatitis C no respondedor a tratamiento previo y con carga viral alta el diagnóstico final fue retrasado debido a una sospecha directamente relacionada con la enfermedad infecciosa. Sin embargo, la respuesta al tratamiento con AUDC fue favorable, como dice la bibliografía médica. En práctica clínica el diagnóstico de LPAC debería sospecharse ante una paciente joven con síntomas biliares (colangitis, ictericia colestásica, cólico biliar) y se debería realizar el análisis genético de ABCB4 para confirmar el diagnóstico. Teniendo en cuenta que, en ocasiones, los pacientes no presentan mutaciones genéticas, este caso supone un ejemplo de la aplicación de la biología molecular en la medicina clínica.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Manuel Romero Gómez
UGMQ Enfermedades Digestivas y CIBERehd
Hospital Universitario de Valme.
Avda. de Bellavista, s/n
41014 Sevilla
e-mail: mromerogomez@us.es
Recibido: 12-03-2014
Aceptado: 31-03-2014
Bibliografía
1. Jacquemin E, De Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, et al. The wide spectrum of multidrug resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001;120:1448-58. [ Links ]
2. Rosmorduc O, Hermelin B, Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 2001;120:1459-67. [ Links ]
3. De Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A 1998;95:282-7. [ Links ]
4. Erlinger S. From MDR3 to LPAC: Cross talk between molecular biology and clinical medicine. Gastroenterol Clin Biol 2010;34:345-6. [ Links ]
5. Fracchia M, Pellegrino S, Secreto P, Gallo L, Masoero G, Pera A, et al. Biliary lipid composition in cholesterol microlithiasis. Gut 2001;48:702-6. [ Links ]
6. Poupon R, Rosmorduc O, Boëlle PY, Chrétien Y, Corpechot C, Chazouillères O, et al. Genotype-phenotype relationships in the low-phospholipid-associated cholelithiasis syndrome: A study of 156 consecutive patients. Hepatology 2013;58:1105-10. [ Links ]
7. Shoda J, Oda K, Suzuki H, Sugiyama Y, Ito K, Cohen DE, et al. Etiologic significance of defects in cholesterol, phospholipid, and bile acid metabolism in the liver of patients with intrahepatic calculi. Hepatology 2001;33:1194-205. [ Links ]
8. Rosmorduc O, Poupon R. Low phospholipid associated cholelithiasis: Association with mutation in the MDR3/ABCB4 gene. Orphanet J Rare Dis 2007;2:29. [ Links ]
9. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:e57. [ Links ]
10. Pasmant E, Goussard P, Baranes L, Laurendeau I, Quentin S, Ponsot P, et al. First description of ABCB4 gene deletions in familial low phospholipid-associated cholelithiasis and oral contraceptives-induced cholestasis. Eur J Hum Genet 2012;20:277-82. [ Links ]
11. Rosmorduc O, Hermelin B, Boelle PY, Parc R, Taboury J, Poupon R. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 2003;125:452-9. [ Links ]
12. Benzimra J, Derhy S, Rosmorduc O, Menu Y, Poupon R, Arrivé L. Hepatobiliary anomalies associated with ABCB4/MDR3 deficiency in adults: A pictorial essay. Insights Imaging 2013;4:331-8. [ Links ]
13. Condat B, Zanditenas D, Barbu V, Hauuy MP, Parfait B, El Naggar A, et al. Prevalence of low phospholipid-associated cholelithiasis in young female patients. Dig Liver Dis 2013;45:915-9. [ Links ]

