










 Custom services
Custom services
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Celiac disease (CD) is a systemic disorder characterized by a variable combination of gluten-dependent clinical manifestations, enteropathy, specific antibodies and HLA-DQ2 and/or HLA-DQ8 genetics 1. Although CD is a multifactorial and polygenic disorder, the main genetic predisposing factors lie within the HLA region. They encode the HLA-DQ2 and/or HLA-DQ8 heterodimers that are present on the surface of antigen presenting cells and involved in CD pathogenesis 2. These receptors are considered as necessary, although not sufficient to develop CD, which confers a high negative predictive value to HLA genotyping 3,4. This was adopted by the new European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) guidelines for CD diagnosis in 2012 1. HLA genotyping is the first CD screening tool in at-risk asymptomatic children and adolescents that is used to select only HLA-DQ2/DQ8 individuals for further CD-specific antibody testing. Genotyping was also proposed to avoid performing biopsies in children with symptoms suggestive of CD with high antibody titers. Nonetheless, it has been recently shown that confirmation with HLA is not necessary in these cases and the recommendation is to restrict HLA typing to patients with intermediate antibody levels in order to rule out false-positive results 5. In any case, HLA is highly relevant in current CD diagnostic guidelines.
HLA-DQ receptors are composed by two subunits, α and β, encoded by two different genes, HLA-DQA1 and HLA-DQB1, respectively. The HLA-DQ2 receptor can be encoded in the cis configuration by the HLA-DQ2.5 haplotype, designated herein as DQ2.5, as HLA will not be used to designate haplotypes in order to distinguish them from HLA-DQ receptors. This haplotype is commonly referred to as DR3-DQ2 and composed by the HLA-DQA1*05:01-HLA-DQB1*02:01 alleles. In the trans configuration, this receptor is encoded by the DQ2.2 and DQ7.5 haplotypes, which are also referred to as DR7-DQ2 and DR5/6-DQ7, respectively, and defined by the HLA-DQA1*02:01-HLA-DQB1*02:02 (DQ2.2) and HLA-DQA1*05:05-HLA-DQB1*03:01 (DQ7.5) alleles. The HLA-DQ8 receptor is encoded by the DQ8 haplotype, which is also referred to as DR4-DQ8 and is composed by the HLA-DQA1*03-HLA-DQB1*03:02 alleles. Around 90% of patients are thought to carry HLA-DQ2 and the majority of the remaining patients have HLA-DQ8. In most cases that lack both of them, only one of the two HLA-DQ2-encoding alleles is present 6, although this is not very relevant for diagnosis. Since the first observations that demonstrated the association of HLA-DQ2 and HLA-DQ8 with CD, many genetic studies have confirmed the role of these heterodimers in the disease, especially in European populations. However, CD risk conferred by HLA genotypes varies across European populations, even though the different HLA allele distributions according to the geographical region are taken into account 7,8. The HLA-DQ distribution and the associated risk of CD development in a pediatric series of CD patients recruited in a single center of Spain was investigated in the present study.
PATIENTS AND METHODS
Design
A retrospective observational case-control study was performed by comparing HLA-DQ data from patients diagnosed with CD at the Hospital Universitario Infantil La Paz (Madrid, Spain) with those from control individuals collected in the same geographical area.
Study subjects
Subjects with CD were selected via a retrospective review of the complete medical records of all patients with HLA-DQ data with suspected CD at the Gastroenterology Department of the Hospital La Paz from 1977 to 2011. CD diagnosis was performed according to the ESPGHAN criteria 9,10. Only patients of a pediatric age (0-16 years old) with Spanish ancestry were included. Patients who maintained a diagnosis of potential CD or without confirmed CD when the medical records were reviewed were excluded. In 2011, 598 subjects with HLA-DQ genotyping had been studied at the department; 475 patients were finally selected, with ages ranging from seven months to 14 years and a mean of 2.6 ± 0.1 years (Fig. 1). The control group consisted of 628 unrelated individuals without immune-related diseases. They were blood-donors, hospital staff or healthy volunteers who gave informed consent before being included in the study. Subjects and controls were unrelated. The study was approved by the corresponding Ethics Committee.

HLA genotyping and nomenclature
DNA was extracted from fresh peripheral blood leukocytes using a "salting out" procedure 11. HLA genotyping was performed using the polymerase chain reaction-sequence specific oligonucleotide probe (PCR-SSOP) method for HLA-DRB1 (HLA-DQA1 and HLA-DQB1).
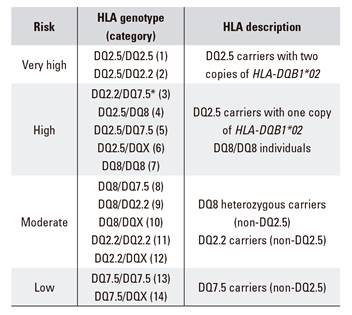
Initially, 15 HLA categories were considered according to the different combinations of the HLA risk haplotypes: DQ2.5, DQ8, DQ2.2 and DQ7.5 as well as non-risk haplotypes: DQX. Thus, carriers of HLA-DQ2.5 were considered, distinguishing between those with two copies of HLA-DQB1*02, i.e. double dose (DQ2.5/DQ2.5 [1] and DQ2.5/DQ2.2 [2]) and those with one copy of HLA-DQB1*02, i.e. single dose (DQ2.2/DQ7.5 or DQ2.5 in the trans configuration [3] and DQ2.5 in the cis configuration, including DQ2.5/DQ8 [4], DQ2.5/DQ7.5 [5] and DQ2.5/DQX [6]). Carriers of HLA-DQ8 were included, distinguishing between homozygous (DQ8/DQ8 [7]) and heterozygous (including DQ8/DQ2.2 [8], DQ8/DQ7.5 [9] and DQ8/DQX [10]). Carriers of HLA-DQ2.2 were also classified according to homozygosity (DQ2.2/DQ2.2 [11]) or heterozygosity (DQ2.2/DQX [12]). Carriers of HLA-DQ7.5 were also included, distinguishing between a homozygous (DQ7.5/DQ7.5 [13]) and heterozygous (DQ7.5/DQX [14]) status and non-carriers of risk heterodimers (DQX/DQX [15]).
Statistical analysis
Genotypic frequencies between cases and controls were compared using the Chi-squared test or Fisher's exact test when appropriate, with 2 x 2 contingency tables. Significant associations were considered as those with p values below 0.05. Odds ratio (OR) with 95% confidence intervals (CI) were used to estimate the relative effect size. Analyses were performed with the EpiInfo v.5.00 statistical package (CDC, Atlanta, USA).
The risk of CD according to the HLA category (R) and prevalence was calculated using the Bayes' rule, which relates different conditional probabilities. In calculation 1,

where "population R" was considered as 1%, the commonly accepted prevalence of CD 12,13. "Sample R" was calculated as the ratio of the number of CD patients to the number of controls for each specific HLA category, and the "sample bias R" was calculated as the ratio of the total number of patients to the total number of controls.
Risks were expressed as odds (1:N), i.e., for each celiac case with that specific HLA genotype there are N individuals with the same genotype without the disease.
RESULTS
In our series of 475 CD patients, 96% carried the HLA-DQ2.5 heterodimer, in contrast to 30% of controls (Table 1). This corroborates the high risk increment conferred by HLA-DQ2.5 (OR = 56.17). Within these HLA-DQ2.5 carriers, a different risk was observed depending on the specific genotype present. The highest risk (OR = 13.76) was conferred by the presence of two copies of the HLA-DQB1*02 allele, which can be observed either in individuals homozygous for the DQ2.5 haplotype or in those with the DQ2.5/DQ2.2 genotype. No differences were observed between these two genetic categories (1 and 2) (OR = 0.95; 95% CI: 0.38-2.39; p = 0.90). With almost a 4-fold lower risk, HLA-DQ2.5 subjects with a single copy of HLA-DQB1*02 were found either in the cis or trans configuration, which combined had an OR of 3.66. No significant differences were observed between the four categories (3, 4, 5 and 6) included in this second risk group.
Table 1 HLA-DQ genotypic frequencies in 475 celiac disease (CD) patients and 628 controls

Double and single dose are used to indicate the presence of two or one HLA-DBQ1*02 alleles, respectively. *DQ2.5 trans; DQX indicates "no risk".
When HLA-DQ2.5 individuals were excluded, 53% of the patients carried the DQ8 haplotype, which appeared in only 18% of the controls. Therefore, within patients that were negative for HLA-DQ2.5, the presence of DQ8 implies an OR of 5.0 (p = 0.0010). A dose effect was also observed as DQ8 in homozygosity confers a significantly higher risk than when in heterozygosity (OR = 33.86; 95% CI: 2.18-1,797.60; p = 0.0038). No significant differences were observed between the high risk observed in DQ8/DQ8 individuals and the risk caused by carrying HLA-DQ2.5 with one (p = 0.44, OR = 0.46) or two copies (p = 1, OR = 1.81) of HLA-DQB1*02. The risk conferred by the presence of the DQ8 haplotype in heterozygosity does not vary significantly according to the presence of additional risk factors (DQ2.2, DQ7.5 or non-risk haplotypes). Although the number of patients in each category is very low and these comparisons lack statistical power.
Carriers of the DQ2.2 haplotype (in absence of any other risk haplotype) represent the second most frequent group among CD patients negative for HLA-DQ2.5, appearing in 37% of cases and only 22% of controls. No dose effect was observed with respect to DQ2.2, as no homozygous DQ2.2 subjects were found in our CD series. No significant differences were observed between the risk conferred by presenting DQ2.2 or presenting DQ8 in heterozygosity (OR = 1.24; 95% CI: 0.37-4.14; p = 0.70).
Finally, the DQ7.5 haplotype appears in the remaining 11% of non-HLA-DQ2.5 carriers (two CD individuals) compared to 27% of the controls. This indicates a 5-fold lower risk compared to the presence of DQ8 or DQ2.2 in heterozygosity (OR = 4.63; 95% CI: 1.03-42.54; p = 0.029). When excluding all the known risk haplotypes (DQ2.5, DQ8 and DQ2.2), DQ7.5 appears in 100% of CD patients but in 44.7% of controls (p = 0.20).
Table 2 Increasing associated celiac disease risk for each genetic category

A prevalence of celiac disease of 1% was considered for the calculations. *DQ2.5 trans; DQX indicates "no risk".
Risks (odds) were calculated using the Bayes' rule for every genetic category and for a prevalence of CD of 1% (Table 2). CD risks ranged from 1:12 to 1:3857. As expected, the highest risk (1:12) was presented by HLA-DQ2.5 carriers with two copies of HLA-DQB1*02. Among HLA-DQ2.5 carriers with a single copy of this allele, the risk of developing CD ranged from 1:35 in carriers of HLA-DQ2.5 inherited in the trans configuration to 1:72 in those with DQ2.5 in combination with DQ8. However, these differences are not significant and the presence of HLA-DQ2.5 in trans or in the different categories including HLA-DQ2.5 in the cis configuration are similar in terms of risk. In individuals who do not carry HLA-DQ2.5, risks are considerably lower and are below the 1:100 general prevalence, 1:854 for heterozygous DQ8, 1:929 for DQ2.2 carriers (excluding those with also DQ2.5, DQ8 and DQ7.5) and 1:3857 for DQ7.5 (in absence of the other risk haplotypes). The only exception is the risk conferred by homozygosity of DQ8, which is 1:25. Based on these results, four categories of genetic risk can be established (Table 3).
DISCUSSION
The involvement of HLA in CD was first described in the 1970's 14,15). Over subsequent years, the specific alleles that underlie the described associations as well as the functional rationale of these observations became clear. HLA-DQ2 and HLA-DQ8 heterodimers show a preference for the presentation of deamidated gluten peptides to CD4+ T cells, triggering the immunological response that characterizes CD 16,17. With the advent of genetic studies, mainly in European populations, the high negative predictive value of HLA-DQ became evident. In 2012, the role of HLA in the diagnosis of CD was firmly recognized 1, which resulted in important changes in diagnostic criteria. It is clear that CD rarely develops in the absence of HLA-DQ2/DQ8. However, a gradient in the HLA frequency exists across Europe and different HLA risk levels of CD have been described 7). Therefore, a detailed analysis in specific populations is required.
Four hundred and seventy-five CD patients that were diagnosed and followed-up in a single center were studied and their HLA-DQ frequencies compared with those present in a sample of 628 ethnically matched unrelated controls. As expected, 96% of patients carried HLA-DQ2.5. CD patients carrying HLA-DQ8 (non-HLA-DQ2.5) were the second most frequent group (2%). Although, HLA-DQ2.2 carriers appear almost at the same frequency (1.5%), mainly considering that two heterozygous DQ8/DQ2.2 are included as HLA-DQ8 carriers. The remaining two patients carried HLA-DQ7.5 (0.4%). The lack of any risk haplotype was the most frequent group in the control population (33%), followed by carriers of HLA-DQ2.5 (30%), HLA-DQ7.5 (27%), HLA-DQ2.2 (22%) and finally HLA-DQ8 (18%). Case-control comparisons showed that the highest risk was present in HLA-DQ2.5 carriers, especially in those with two copies of the HLA-DQB1*02 allele. This has been consistently observed previously 18,19,20,21. A dose effect was also observed in relation to DQ8. Although the low number of DQ8 homozygous individuals precludes us from determining the exact risk level of this genotype. However, it is probably an intermediate level between the two risk categories of HLA-DQ2.5 (with one or two copies of HLA-DQB1*02). The double dose effect of DQ8 has been previously described in some populations 6. More noticeable is the similar risk conferred by DQ8 and DQ2.2 in non-HLA-DQ2.5 in our cohort. Importantly, two children that only carried the DQ7.5 haplotype were identified that are commonly considered as conferring no risk.
Similar low frequencies of DQ8 have been reported in children with CD from other Spanish regions 22,23,24,25. In addition, the frequencies observed in our Spanish controls differ from those reported in the North of Europe and are slightly lower for DQ2.5. Although they are higher for DQ2.2 and DQ7.5, as described in other southern European populations (7,8). The HLA-DQ frequencies observed in CD patients will be concordant with the HLA composition of the studied population. Therefore, if these confer a risk, it is not surprising to find a higher representation of DQ2.2 and DQ7.5 among our CD cohort. This could also contribute to the high proportion of DQ7.5 CD patients described in Italy, where a frequency of 28% was reported for the DQ7.5 haplotype 26.
The risk to develop CD is influenced by the specific gluten peptides presented to CD4+ T cells and the quantity 27, and will also depend on the risk haplotype present, including DQ7.5. In addition, it must be considered that other HLA and/or non-HLA risk factors contribute to CD heritability and could be modulating the observed risk.
Considering a prevalence of 1:100, the odds of having CD was calculated according to the HLA-DQ genotype. A value of 1:31 was found for HLA-DQ2.5 carriers, which rose to 1:12 when two copies of HLA-DQB1*02 were present. Odds of 1:46 was observed in carriers of one copy and this varied widely depending on the other haplotype present, although no significant differences were found for DQ2.5 heterozygosity with DQ2.2, DQ7.5 and non-risk haplotypes. This is quite unexpected considering the expanded peptide-binding repertoire present in carriers of two different risk haplotypes, which should lead to increased CD4+ T cell stimulation. It is even more striking when considering that the highest odds (1:42) was observed in HLA-DQ2.5 carriers with no additional risk haplotypes, followed by those carrying DQ7.5 (1:60) and patients with DQ8 (1:72). However, this result can be interpreted according to the observations of Lenz et al. 28. These authors observed non-additive effects in the HLA region. Specifically, an interaction between DQ2.5 was found with three other haplotypes; from highest to lowest intensity these were DQ2.2, DQA1*01:01-HLA-DQB1*05:01 and DQ7.5. It is interesting that an interaction was not observed with DQ8, but with a haplotype included in the "non-risk haplotypes". In our cohort, 33% of DQ2.5/DQX patients carried the haplotype DQA1*01:01-HLA-DQB1*05:01, which was observed in 26% of DQ2.5/DQX controls.
When HLA-DQ2.5 carriers were excluded, an odds higher than the disease prevalence was only observed for DQ8 homozygosity. The presence of DQ8 (excluding homozygous individuals), DQ2.2 and DQ7.5 implies odds of 1:854, 1:1059 and 1:3857, respectively. No individuals that lacked all susceptibility alleles were found in our sample. This last observation highlights the relevance of considering all risk haplotypes when diagnosing CD.
In summary, based on our results, four groups of CD risk can be established. The first two groups comprise HLA-DQ2.5 carriers and DQ8 homozygous subjects and show an increased risk compared to the general population. The third group comprises DQ8 heterozygotes and DQ2.2 carriers. Although DQ8 is undisputed, conflicting data are found in the literature with regard to DQ2.2, as the risk of the HLA-DQB1*02 allele is commonly considered to be restricted to the co-existence with HLA-DQA1*05. Finally, a fourth group can be defined that includes individuals with DQ7.5. Even though a significant result was not found when the frequency of DQ7.5 was compared between cases and controls, DQ7.5 is present in all patients that are not included in the aforementioned groups. This has also been observed in different countries, including Spain 6,29. In Italy, DQ7 is thought to represent an additive or independent CD risk haplotype 26. Therefore, it is important that this haplotype is considered when a high suspicion of CD exists before ruling-out CD based on HLA genotyping.
These results may have an important impact in the clinical practice. Especially when developing personalized programs focused on subclinical CD in relatives or children of other risk groups and to define the timing of serological screening depending on the HLA-DQ genotype.
