










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
Print version ISSN 1137-6627
Anales Sis San Navarra vol.26 n.1 Pamplona Jan./Apr. 2003
ARTÍCULOS ORIGINALES
Evaluación de la práctica clínica del Consentimiento Informado en los ensayos clínicos
Evaluation of the clinical practice of Informed Consent in clinical trials
J. Gost1, C. Silvestre2, P Ezpeleta3, P. Astier2, O. Díaz de Rada1, M.T. Artázcoz4
|
RESUMEN Sobre la base de las publicaciones existentes parece lícito suponer que en un ensayo clínico (EC), las dificultades inherentes al proceso de comunicación investigador-participante son en la práctica mayores que las deseadas. Se asume asimismo la hipótesis de que existen deficiencias en la legibilidad formal de los documentos de consentimiento informado.
|
ABSTRACT On the basis of existing publications it would seem legitimate to assume that in a clinical test (CT) the difficulties inherent in the process of researcher-participant communication are in practice greater than desired. Similarly, the hypothesis is adopted that difficulties exist in the formal legibility of the documents of Informed Consent. Key words. Clinical trials with medicines. Informed consent. Legibility. Helsinki Declaration. Ethical Committee of Clinical Research. |
| 1. Comité Etico de Investigación Clínica de Navarra. 2. Servicio de Medicina Preventiva y Gestión de Calidad. Hospital de Navarra. Servicio Navarro de Salud-Osasunbidea. 3. Becaria con cargo al proyecto. 4. Departamento de Salud. Gobierno de Navarra. + Proyecto realizado con financiación del Departamento de Salud del Gobierno de Navarra. Premio Mikel Larumbe 2000 Aceptado para su publicación el 13 de febrero de 2003. | Correspondencia Javier Gost Garde Comité Etico de Investigación Clínica de Navarra Pabellón de Docencia Hospital de Navarra Irunlarrea, 3 31008 Pamplona E-mail: jgostgar@cfnavarra.es |
INTRODUCCIÓN
Marco legislativo
El Real Decreto (RD) 561/1993, de 16 de abril, establece los requisitos para la realización de ensayos clínicos (EC) con medicamentos1.
Dicha norma básica establece en su Artículo 10 que todos los EC habrán de contar, antes de poder ser realizados, con el informe previo del correspondiente Comité Ético de Investigación Clínica (CEIC) y que los mismos se realizarán en condiciones de respeto a los derechos fundamentales de la persona y a los postulados éticos que afectan a la investigación biomédica con seres humanos, siguiendo a estos efectos los principios contenidos en la Declaración de Helsinki (DH) y sucesivas actualizaciones2.
El Artículo 12 define y regula la obtención del consentimiento informado (CI). El CI es el procedimiento que garantiza que el sujeto ha expresado voluntariamente su intención de participar, siendo pues imprescindible que el sujeto otorgue libremente su consentimiento antes de poder ser incluido en un EC.
El Artículo 42 regula las funciones de los CEIC, entre las que cabe destacar –al tener relación directa con este estudio– las que corresponden a:
Evaluar la información escrita sobre las características del ensayo, la forma en que dicha información será proporcionada y el tipo de consentimiento que va a obtenerse.
Realizar el seguimiento del EC desde su inicio hasta la recepción del informe final.
En la Comunidad Foral de Navarra existe un único CEIC para el conjunto de los establecimientos sanitarios ubicados en la misma, y su constitución y funciones vienen reguladas mediante Decreto Foral3.
De los antecedentes mencionados se deduce que la principal función de los CEIC es la de garantizar la protección de las personas que participan en un EC. Para ello, el CEIC se basa en el estudio de dos tipos de documentación diferente: el protocolo del EC (incluida la cualificación del equipo investigador) y el de la información que sobre el desarrollo del EC se facilita a los sujetos (documento de información y consentimiento informado).
Por lo que respecta al protocolo del EC (su pertinencia, diseño y metodología) existe evidencia suficiente para afirmar que su calidad ha mejorado en el transcurso de los años, y que, en parte, también ha sido debido a la labor de los comités éticos4. Sin embargo, los CEIC disponen de una escasa información acerca de las circunstancias en que se desarrolla cada EC. No hay que ignorar que, a pesar de la existencia de normas de buena práctica clínica y de los mecanismos instaurados para garantizar su calidad, existe una amplia bibliografía sobre errores y fraudes en los EC5-8.
Con relación a la práctica del consentimiento informado existen dificultades objetivas9-12, descritas ya hace más de quince años, ligadas al propio desarrollo de la relación médico-paciente y que provienen tanto del conflicto de intereses como por la existencia de una relación de dependencia entre ambos actores, que puede condicionar, especialmente si la investigación se realiza en el contexto de la asistencia sanitaria, la participación del paciente en el EC.
Si ya de por sí resulta complejo encontrar cuál es el mejor método para la obtención del consentimiento informado en los EC13-15, todavía lo es más verificar si ha existido un intercambio de información que minimice los problemas inherentes a dicho proceso o, si por el contrario, se ha limitado a cumplir con un trámite legal, que se resume en la entrega de un documento repleto de terminología médica especializada y que carece de un proceso de validación previo por lo que su comprensibilidad puede verse seriamente comprometida16-20.
Siendo conscientes de las diferencias que pueden existir entre el diseño teórico y la práctica del CI en los ensayos clínicos21,22 y teniendo en cuenta que la experiencia previa nos indica que tanto los órganos competentes de la autoridad sanitaria como los propios CEIC desarrollan una escasa capacidad operativa para ejercer, respectivamente, las competencias que en materia de inspección y seguimiento tienen encomendados, surge la pregunta clave: ¿qué evidencias existen de que los requisitos regulados por la citada normativa se cumplen durante el desarrollo del EC?
Para intentar responder a este interrogante se diseñó este estudio con los siguientes objetivos:
Evaluar la existencia del documento de Consentimiento Informado en los ensayos clínicos informados por el CEIC de Navarra durante el período 1995-99.
Evaluar si el contenido del documento de información para los participantes presentado para su revisión por el CEIC responde a lo previsto en el Real Decreto 561/93 y a las directrices de la Declaración de Helsinki.
Detectar áreas de mejora y proponer soluciones a las mismas.
Evaluar si el documento de información escrita presentado a los participantes coincide con el presentado para su aprobación al CEIC.
Conocer el grado de legibilidad formal de los documentos informativos facilitados a los participantes en los EC.
METODOLOGÍA Y PLAN DE TRABAJO
La fuente de datos la han constituido la base de datos y archivos del CEIC de Navarra.
Diseño del estudio: transversal
Población de muestreo: la unidad de muestreo es el ensayo clínico. El CEIC en el período 1995-99 ha informado positivamente 289 EC. El tamaño de la muestra (n = 160), se ha calculado sobre la hipótesis de que al menos un 12% de los EC presentarán algún tipo de defecto formal en el cumplimiento del CI, con un peor esperado de ± 4 puntos, nivel confianza del 95% y pérdidas del 20%. La selección de la muestra fue aleatoria.
Variables del EC estudiadas: centro de realización, investigador, fase, diseño metodológico (aleatorización, enmascaramiento, etc.), existencia de cuaderno de recogida de datos (CRD). Con respecto al documento de CI: existencia en archivo, coincidente con el presentado para su aprobación por el CEIC, firma del participante, firma del investigador, contenido de las áreas relevantes (objetivos, metodología y protección de los derechos del participante), complejidad oracional y de vocabulario e índices derivados.
– Criterios utilizados en variables dicotómicas (SÍ/NO). La ausencia de documentación en archivos ha sido valorada como no realización de la variable investigada (v.g. existencia de documento de CI). Asimismo, la ausencia de la firma del investigador y/o del participante en un solo documento de CI (con independencia del número de participantes en un ensayo concreto) ha sido valorada como no realización, ya que la ley obliga a cumplimentar todos los documentos de CI y resulta suficiente que un solo documento de CI esté sin firmar en un EC para valorarlo como no realizado.
Procedimiento. Obtención de la muestra de EC. Comunicación a todos los investigadores seleccionados y directores de los centros de la realización del estudio. Entrevista con los investigadores. Revisión de la documentación y recogida de datos.
Herramienta de legibilidad de Microsoft Word para Windows NT. Al aplicar la herramienta se ha intentado eliminar los sesgos sistemáticos asociados al documento informativo; por ello se ha realizado una revisión de cada texto, adaptándolo a las normas de corrección, y se ha aplicado exclusivamente al texto informativo, despojado de artefactos v.g. título del EC, Dr., Estimado Sr., etc. Los documentos analizados han sido los presentados por el promotor al CEIC para su evaluación y corresponden a los EC de la muestra.
Análisis de datos. El análisis estadístico se ha realizado mediante SPSS 10.0. Las variables cuantitativas se describen con la media y la desviación estándar; las cualitativas, con la distribución de porcentajes de cada una de las categorías. El estudio de la asociación entre variables cualitativas se ha realizado mediante la prueba de la c2. El nivel de significación estadística aceptado ha sido del 5%.
Análisis de legibilidad:
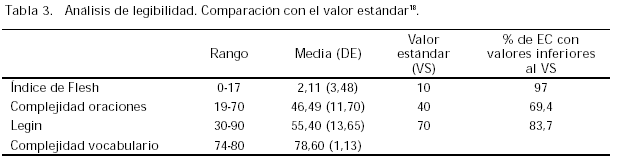
– Índice de Flesch (IF): este número es alto cuando el documento resulta fácil de leer y bajo cuando la redacción es complicada; su valor varía de 0-100 y se obtiene de acuerdo a un estándar norteamericano23.
– La complejidad oracional (CO: oraciones demasiado largas o con una estructura muy complicada) y del vocabulario (palabras demasiado largas o inusuales) varían entre 0-100 y su valoración es alta cuando son complejas.
– El índice Legin, que es una combinación de los anteriores, varía también entre 0-100 y se comporta de manera semejante al Flesch. Se calcula según la fórmula 100+IF-CO.
RESULTADOS
La distribución por fase y centro de realización de los 160 EC muestreados es la que figura en la tabla 1. La Fase del EC se corresponde con las definidas en el Real Decreto 561/93. Los hospitales A, B y C son hospitales de nivel terciario. El apartado otros centros agrupa al resto de establecimientos sanitarios en los que se ha realizado al menos un EC.

Por lo que respecta al diseño de los EC, el 80% eran aleatorizados (128 ensayos), el 81,2% tenían grupo control (130 ensayos), contaban con algún tipo de enmascaramiento el 45% (72 ensayos) y eran estudios con placebo el 20,6% (33 ensayos).
De los 160 EC muestreados se han realizado 122, lo que supone que 38 ensayos (23,8%) a pesar de haber sido informados favorablemente por el CEIC no han sido llevados a la práctica. Las causas más habituales corresponden a haber sido retirados por el promotor, no haber llegado a un acuerdo económico o por no haber sido autorizado por la dirección del centro. Los porcentajes de distribución por centros de realización y fase del EC en los 122 ensayos realizados, son muy similares a los encontrados para el total de la muestra, no existiendo diferencias significativas entre los mismos.
De los 122 EC realizados, sólo en un 69,7% (85 casos) el investigador archivaba los datos correspondientes a dicho ensayo. A los efectos del análisis de datos, la ausencia de prueba se ha considerado como no realización del ítem evaluado, por lo que solamente se ha considerado que existe documento de CI en el 69,7% de los EC.
En aquellos ensayos en los que se archivan datos (85 casos), la firma del paciente figuraba en el 100% de los documentos de CI, mientras que la del investigador aparecía sólo en el 81,25% (69 casos). La información escrita era coincidente con la presentada ante el CEIC en el 95,3% de los casos.
Teniendo en cuenta que la ausencia de prueba ha sido considerada como no realización del CI, esto significa que en el conjunto de los 122 EC realizados, el documento de CI aparecía firmado por el paciente en el 69, 7% de los ensayos y estaba firmado por el investigador en tan sólo un 56,6% de los ensayos.
Los resultados del contenido de la información que se facilita a los participantes figuran en la tabla 2. Los resultados del estudio de legibilidad formal figuran en la tabla 3. El análisis de la información escrita se ha efectuado sobre el total de la muestra seleccionada (160 documentos de CI) ya que al estar constituida la fuente de datos por los archivos del CEIC no ha habido ninguna pérdida en este caso.

No se han encontrado asociaciones estadísticamente significativas entre las variables dependientes principales (existencia de archivos, firma del documento de CI, legibilidad) y el resto de variables independientes (centro de realización, fase del EC, etc.). Cuando se estudia la relación entre el hospital donde se realiza el EC y el mantenimiento por parte de los investigadores de archivos específicos para los EC, las cifras se encuentran próximas a la significación estadística (p < 0,06) para un determinado centro.
El escaso número de EC asociados a un determinado investigador y/o promotor no permite la valoración asociada a dichas variables.
DISCUSIÓN
En primer lugar hay que destacar el elevado porcentaje (23,8%) de EC no realizados en el conjunto de la muestra analizada. Aunque el porcentaje de pérdidas es ligeramente superior al estimado al realizar el cálculo muestral, entendemos que no afecta a la validez global de los datos obtenidos dado el nivel de confianza asumido. Sin embargo, es un dato preocupante, por cuanto indica que el trabajo del CEIC es claramente ineficiente. Dedica una gran cantidad de tiempo a evaluar unos ensayos clínicos que posteriormente no se llevan a la práctica debido fundamentalmente a una deficiente coordinación entre los diferentes actores del proceso (promotor-investigador-dirección del centro).
En aproximadamente una tercera parte de los EC realizados el investigador no ha conservado la documentación correspondiente. Por parte del equipo investigador se ha debatido si la ausencia de archivos en un porcentaje tan elevado de ensayos clínicos pudiera corresponder a una falta de colaboración de los investigadores, que adoptara el pretexto de no haber archivado los datos. Aunque inicialmente se detectaron reticencias en algunos investigadores, las mismas desaparecieron cuando se explicaron personalmente los objetivos del estudio, por lo que creemos que realmente nos encontramos ante una inexistencia de archivos. Cuando se estudia la relación entre el hospital donde se realiza el EC y el mantenimiento de archivos específicos para los EC, el hecho de que para un determinado centro las cifras se encuentren próximas a la significación estadística entendemos que debe ser tomado en consideración, máxime cuando el reducido tamaño de la muestra está condicionando su eventual significación. El que en uno de los tres hospitales estudiados el porcentaje de EC en los que no se guarda ningún tipo de documentación sea más del doble que en el resto de los hospitales, nos indica una importante área de mejora.
Asimismo, el equipo investigador ha adoptado unos criterios estrictos (ver metodología) para considerar como positivo el cumplimiento de determinadas variables (v.g. documento firmado de CI). Es posible que estemos magnificando el grado de incumplimiento (además de modificar al alza la hipótesis de partida) y que realmente el proceso de información investigador-paciente se haya producido, pero la ausencia de prueba en todos y cada uno de los casos obliga a valorarla como no realizada.
La valoración de la legibilidad es una tarea compleja, tanto por el propio proceso como por las herramientas existentes, dado que las mismas no están, salvo excepciones, validadas al castellano18,24. En este estudio se ha optado por una herramienta automática y posterior cálculo del índice Legin. Aun cuando se ha intentado evitar los sesgos sistemáticos, la casi nula información facilitada por el distribuidor de la herramienta (Microsoft) sobre sus normas internas de aplicación así como su falta de validación al castellano, obliga a ser prudente en la interpretación de los datos obtenidos. Con todo, los resultados, además de ser coincidentes con los encontrados en otros estudios18, se asemejan más a los calculados para las revistas biomédicas que a las publicaciones de información general, tal y como era de esperar dado el tipo de información facilitada.
Dada la metodología del estudio, los datos son de aplicación en el ámbito de la Comunidad Foral Navarra. Sin embargo, el hecho de que los centros sanitarios de Navarra, tanto del sector público como del privado, participen en un elevado número de ensayos multicéntricos, hace pensar que los resultados obtenidos en el apartado de contenidos y legibilidad de la información, pudieran ser indicativos de lo que sucede en el ámbito estatal, por lo que se sugiere la conveniencia de realizar estudios similares que garanticen la representatividad del universo de todos los EC revisados en España.
CONCLUSIONES
Aproximadamente uno de cada cuatro EC informados por el CEIC no se llevaron finalmente a la práctica.
En una tercera parte de los realizados no se conservaba la documentación correspondiente al ensayo.
Aun cuando no se han encontrado diferencias estadísticamente significativas en relación con la existencia o no de archivos de los EC entre los centros A, B y C, las diferencias encontradas con respecto a uno de los centros orientan a realizar una acción de mejora entre los investigadores adscritos a dicho centro.
En aquellos EC en los que se conservaban datos, la firma del investigador sólo estaba presente en el 80 % de los documentos de CI (la ausencia de la firma del investigador en un solo CI, con independencia del número de participantes en un ensayo concreto, ha sido valorada como no realización).
En aquellos ensayos en los cuales existe documento de Consentimiento Informado, casi un 5% de dichos documentos no coinciden con los presentados al CEIC para su revisión.
El documento de consentimiento informado enviado a revisión por el promotor responde, en términos generales, a los principios recogidos tanto en la Declaración de Helsinki como en la legislación española. El grado de ajuste es superior al 90% en buena parte de los apartados. Las áreas prioritarias cuya información debe ser mejorada se corresponden prioritariamente con el área de los derechos del paciente y, entre otras, cabe citar:
– Posibilidad de interrupción por el participante.
– Cómo y/o dónde contactar con el investigador.
– Revisión del ensayo por parte del CEIC.
La legibilidad formal de los consentimientos informados facilitados a los participantes en EC es muy deficiente. La complejidad del vocabulario es muy elevada. Si la información facilitada se realizase fundamentalmente sobre la base de la documentación escrita, las dificultades del sujeto participante para comprender la información y prestar su consentimiento serían importantes.
Las implicaciones prácticas detectadas figuran enunciadas en la tabla 4.

BIBLIOGRAFÍA
1. Boletín Oficial del Estado. Real Decreto 561/1993 por el que se establecen los requisitos para la realización de ensayos clínicos con medicamentos. BOE núm 114, de 16-04-1993. [ Links ]
2. La Declaración de Helsinki: http://www.wma.net/e/policy/17-c_e.html. [ Links ]
3. Boletín Oficial de Navarra. Decreto Foral 252/1996, de 24 de Junio, por el que se modifica la composición del Comité Etico de Investigación Clínica de la Comunidad Foral de Navarra. BON nº 84, de 12-07-1996. [ Links ]
4. Savulescu J, Chalmers I, Blunt J. Are research ethics committees behaving unethically? Some sugestions for improving performance and accountability. Br Med J 1996; 313: 1390-1393. [ Links ]
5. Research misconduct: a resumé of recent events. Lock S. Fraud and misconduct in medical research. Lock S, Wells F, editores. London: Br Med J Publishing 1993; 5-24. [ Links ]
6. International Conference on Harmonization of Technical Requeriments for the Registration of Pharmaceutical for Human Use (ICH). Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95). 01 May 1995. [ Links ]
7. Vallvé C. La buena práctica clínica y la caja de Pandora (I). Los comités éticos de investigación clínica. Med Clin (Barc) 1996; 107: 657-659. [ Links ]
8. Vallvé C. La buena práctica clínica y la caja de Pandora (y III). La fiabilidad de los datos. Med Clin (Barc) 1997; 108: 65-67. [ Links ]
9. Byrne DJ, Napier A, Cuschieri A. How informed is signed consent? Br Med J 1988; 296: 839-840. [ Links ]
10. Vallvé C. La buena práctica clínica y la caja de Pandora (II).El consentimiento informado. Med Clin (Barc) 1997; 108: 19-22. [ Links ]
11. Guix J, Balañá Ll, Carbonell JM, Simón R, Surroca RM, Nualart Ll. Cumplimiento y percepción del consentimiento informado en un sector sanitario de Cataluña. Rev Esp Salud Pública 1999; 73: 669-675. [ Links ]
12. Simón P, Júdez J. Consentimiento informado. Med Clin 2001; 117: 99-106. [ Links ]
13. Edwards SJ, Lilford RJ, Thornton J, Hewison J. Informed consent for clinical trials: in search of the best method. Soc Sci Med 1998; 47: 1825-1840. [ Links ]
14. Davis TC, Holcombe RF, Berkel HJ, Pramanik S, Divers SG. Informed consent for clinical trials: a comparative study of standar versus simplified forms. J Natl Cancer Inst 1998; 90: 668-674. [ Links ]
15. Verheggen FW, van Wijmen FC. Myth and reality of informed consent in clinical trials. Med Law 1997; 16: 53-59. [ Links ]
16. Ordovás JP, López E, Urbieta E, Torregrosa R, Jiménez N. Análisis de las hojas de información al paciente para la obtención de su consentimiento informado en ensayos clínicos. Med Clin (Barc) 1999; 112: 90-94. [ Links ]
17. Simón Lorda P. Legibilidad de los formularios escritos de consentimiento informado del Servicio Vasco de Salud-Osakidetza. Grupo de trabajo sobre consentimiento informado en Osakidetza. Rev Calidad Asistencial 1999; 14: 95-99. [ Links ]
18. Simón Lorda P, Barrio Cantalejo MI, Concheiro Carro L. Legibilidad de los formularios escritos de consentimiento informado. Med Clin (Barc) 1996; 107: 524-529. [ Links ]
19. Hammerschmidt DE, Keane MA. Institutional Review Board (IRB) review lacks impact on the readability of consent forms for research. Am J Med Sciences 1992; 304: 348-351. [ Links ]
20. Philipson SJ, Doyle MA, Gabram SG, Nightingale C, Philipson EH. Informed Consent for Research: A study to evaluate readability and processability to effect change. J Invest Med 1995; 43: 459-467. [ Links ]
21. Werheggen FW, van Wijmen FC. Informed consent in clinical trials. Health Policy 1996 May; 36: 131-153. [ Links ]
22. Alderson P. Informed consent: ideal or reality? J Health Ser Res Policy 1998; 3: 124-126. [ Links ]
23. En: http://eho.org/pdf/100ln.pdf.
24. Fernández Huerta J. Medidas sencillas de lecturabilidad. Consigna 1959; 214: 29-32. [ Links ]

