










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de Osteoporosis y Metabolismo Mineral
On-line version ISSN 2173-2345Print version ISSN 1889-836X
Rev Osteoporos Metab Miner vol.8 n.4 Madrid Oct./Dec. 2016
EDITORIAL
Chronic renal failure, vascular calcification and the RNK/RANKL/OPG system
Insuficiencia renal crónica, calcificación vascular y sistema RNK/RANKL/OPG
Olmos J.M. and Hernández J.L.
Departamento de Medicina Interna - Hospital Universitario Marqués de Valdecilla-IDIVAL - Universidad de Cantabria - Santander (España)
e-mail: miromj@humv.es
Cardiovascular complications are among the most important clinical challenges in patients with chronic kidney failure (CKF). These are frequent processes that present high morbidity and mortality. As an example, around 50% of patients with terminal CRF die from this disease1.
Renal patients present two types of vascular calcifications: calcification of the tunica media, also called Mönckeberg sclerosis, in which the mineral is deposited within the layer of smooth muscle. The second type is calcification of the intima, in which the calcium deposit occurs after the accumulation of cholesterol under the damaged endothelial monolayer2. Calcification of the tunica media, where vascular smooth muscle cells (VSMC) and elastic fibers are found, is not related to cholesterol levels or the existence of atheromatous plaques and causes the hardening and decrease in the arteries' distensibility. Atherosclerotic calcification of the intima may also occur in patients with CRF. In these cases, intimal calcification is associated with the subintimal deposit of lipids and lipoproteins, which may stimulate the development of immune responses, both innate and adaptive, inducing endothelial cells and the VSMC to express inflammatory molecules, which stimulate tumor-infiltrating monocyte/macrophage.
As a result, increased inflammation, oxidized lipids and fibrous matrix secretion in atherosclerotic lesions further accelerate vascular calcification, which eventually leads to atherosclerotic plaque rupture1-3. In patients with CKF, both atherosclerotic intimal calcification and tunica media calcification, independent of atherosclerosis, are associated with an increase in cardiovascular mortality compared to patients with CKF who do not present it4.
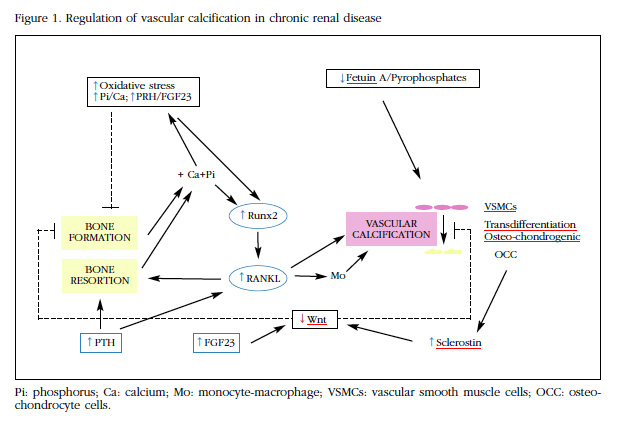
Initially this was considered a secondary disorder to the passive deposit of calcium and phosphorus in the vascular wall. However, more recently vascular calcification has been found to be a perfectly regulated process by which VSMC undergo molecular and phenotypic changes. With these alterations they acquire some of the functions that characterize osteo-chondrocitary strain cells1,2, and lead to the release by vesicular VSMC structures containing hydroxyapatite5. In this process of "osteo-chondrocyte transdifferentiation" different factors involved in the differentiation of bone cells, such as Runx2, bone morphogenic proteins (BMPs), RANK/RANKL/OPG system or Wnt pathway would intervene. Furthermore, in patients with CKF and in animal models of this disease, increased vascular calcification is accompanied by a reduction in bone mass, suggesting that the signals involved in bone and vascular wall mineralization may behave differently depending on the tissue microenvironment in which they act6,7.
CKF is characterized by changes in bone metabolism that, in addition to being detrimental to the skeleton -renal osteodystrophy- favor calcification of soft tissues and vessels. Hypercalcemia and hyperphosphatemia, hyperparathyroidism, increased fibroblast growth factor 23 (FGF23), increased oxidative stress and decreased inhibitors of calcification such as fetuin-A and pyrophosphates could all play a role in the vascular calcification process1,2,6,7.
Hyperphosphatemia, as well as hypercalcemia, are two of the main factors associated with the development of vascular calcification in CRF8. The diet with high phosphorus content increases vascular calcification and reduces bone mass in rats with chronic renal failure. On the other hand, treatments with high calcium and/or phosphate content induce the calcification of VSMC in experimental animals. Although the mechanisms involved in this process have not yet been accurately found, hyperphosphatemia has been shown to induce vascular calcification, favoring osteogenic expression such as Runx2 or BMP29,10. Some authors have shown that, unlike normal vessels, the arteries of CKD patients express Runx21,2,9,10. On the other hand, the uremic serum increases the expression of Runx2 and the calcification of the VSMC. In addition, hyperphosphatemia activates the Wnt pathway, favoring β-catenin translocation in the smooth muscle cell nucleus, thereby stimulating the expression of direct target genes such as cyclin D1, axin 2 and VCAN/versican10. Finally, hyperphosphatemia also increases the levels of FGF23, which, together with its co-receptor klotho, may play a pathogenic role in arterial calcification and in the alteration of skeletal mineralization11.
The role of PTH is also complex. In hemodialysis patients, increased PTH is associated with vascular calcification and, in rats with renal failure, both aortic calcification and loss of bone mass are associated with increased phosphorus and PTH12. However, in other studies it has been pointed out that PTH is not able to directly induce vascular calcification, but would have a synergistic effect with phosphate, which would be related to increased osteoclastic activity and bone remodeling that this hormone determines. This increase in bone remodeling favors calcium and phosphorus loss from the bone, thus stimulating vascular calcification. It is one of the determinants of the most frequent forms of renal osteodystrophy, osteopathy with high remodeling or secondary hyperparathyroidism. At other times, as with adynamic bone, the low bone remodeling determines an alteration in bone formation and mineralization, with the consequent reduced use of excess calcium and phosphorus, which also favors vascular calcification13,14.
The increase in oxidative stress observed in patients with CRF would also be closely associated with the development of vascular calcification. As with hyperphosphatemia, this effect would be mediated through the expression of Runx2 in the VSMC15. In addition, a recent study carried out in postmenopausal women found that increased oxidative stress was associated with an increased risk of hip fracture, suggesting that there would be an inverse relationship between oxidative stress and mineral metabolism1,5.
Along with increased levels of calcium and phosphorus, the decrease in some of the inhibitors of calcification, such as fetuin-A and pyrophosphate, which can be observed, can contribute to the increase of vascular calcification in these patients1,2,6.
In this issue of the Journal of Osteoporosis and Mineral Metabolism, Martínez Arias et al.16 analyzed the effects of the RANK/RANKL/OPG system on bone demineralization and vascular calcification in CRF. These authors use in vivo and in vitro models of vascular calcification to verify that rats with chronic renal failure and a diet high in phosphorus present decreased bone mineral density, together with aortic calcifications that are accompanied by an increase in RANKL gene expression and a decrease in OPG. In the tibia of these animals both RANKL and OPG expression increased, although the increase in OPG occurred at earlier stages. In the VCAM, the addition of uremic serum and calcifying medium induced an increase in calcium content and RANKL and OPG expression, while the addition of OPG and the silencing of RANK inhibited this phenomenon. Therefore, these authors' results confirm the RANK/RANKL/OPG axis involvement in the vascular calcification process and probably also in the loss of bone mass that accompanies CRF. This opens the door to new research lines in this area.
As the authors16 comment, there is a great deal of scientific evidence linking the RANK/RANKL/OPG system to vascular calcifications1,2,7,16-20. The first derivative of the OPG-null mouse study conducted a few years ago by Bucay et al.17, who demonstrated that OPG-deficient mice exhibited vascular calcifications, as well as an intense decrease in bone mineral density (BMD) and one High incidence of fractures. It was later found that treatment with recombinant OPG significantly reduced vascular calcification in mice deficient in LDL receptors18. On the other hand, the studies carried out in patients with CKD indicate that the levels of RANKL and OPG increase as do those of PTH and phosphate, and it has been pointed out that the increase of Runx2 increases the expression of RANKL in VCAM. In animal models, increased RANKL induces a loss of bone mass and vascular calcification, while the addition of OPG has the opposite effect. The pathway by which RAKL would promote calcification would be through binding to its RANK receptor, with the consequent activation of the NF-kB alternative pathway and the bone morphogenic proteins 2 and 4 (BMP2 and BMP4), favoring the osteogenic transition of the VSMC1,2,19,20. On the other hand, RANKL could also act indirectly by stimulating the release of pro-cytokines by macrophages.
Finally, and as might be expected, the Wnt pathway also appears to be involved in this process. We have already commented that hyperphosphatemia would activate this pathway in the VCAM10. On the other hand, the expression of sclerostin increases in arteries with vascular calcification. Levels of sclerostin and other Wnt pathway inhibitors, such as Dickkopf-1 (DKK1) or soluble frizzled receptor (SFR), increase as renal function deteriorates and correlate inversely with histological parameters of bone remodeling and with the number and function of osteoblasts21,22. It has recently been pointed out that the increase of FGF23, which accompanies renal function deterioration, could also act to inhibit this system11. Therefore, sclerostin and other inhibitors of the Wnt system, released into the medium from the vessels, could act to impair the bone structure and retard the mineralization process. These alterations, along with those of the RANK/RANKL/OPG system, hyperphosphatemia and other factors discussed here, could help medical researchers to understand the complex relationship between vascular calcification and bone loss and increased fractures in patients With CRF (Figure 1).
Bibliography
1. Byon CH, Chen Y. Molecular Mechanisms of Vascular Calcification in Chronic Kidney Disease: The Link between Bone and the Vasculature. Curr Osteoporos Rep. 2015;13:206-15. [ Links ]
2. Lu KC, Wu CC, Yen JF, Liu WC. Vascular calcification and renal bone disorders. Scientific World Journal. 2014;2014:637065. [ Links ]
3. Harper E, Forde H, Davenport C, Rochfort KD, Smith D, Cummins PM. Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vascul Pharmacol. 2016; 82:30-40. [ Links ]
4. London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18(9):1731-40. [ Links ]
5. Reynolds JL, Joannides AJ, ↑ JN, McNair R, Schurgers LJ, Proudfoot D, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857-67. [ Links ]
6. Zheng CM, Zheng JQ, Wu CC, Lu CL, Shyu JF, Yung-Ho H, et al. Bone loss in chronic kidney disease: Quantity or quality?. Bone. 2016;87:57-70. [ Links ]
7. Cannata-Andia JB, Roman-Garcia P, Hruska K. The connections between vascular calcification and bone health. Nephrol Dial Transplant. 2011;26:3429-36. [ Links ]
8. Mathew S, Tustison KS, Sugatani T, Chaudhary LR, Rifas L, Hruska KA. The mechanism of phosphorus as a cardiovascular risk factor in CKD. J Am Soc Nephrol. 2008;19:1092-105. [ Links ]
9. Mikhaylova L, Malmquist J, Murminskaya M. Regulation of in vitro vascular calcification by BMP4, VEGF and Wnt3a. Calcif Tissue Int. 2007;81:372-81. [ Links ]
10. Martínez-Moreno JM, Muñoz-Castañeda JR, Herencia C, Oca AM, Estepa JC, Canalejo R, et al. In vascular smooth muscle cells paricalcitol prevents phosphate-induced Wnt/b-catenin activation. Am J Physiol Renal Physiol. 2012;303:F1136-44. [ Links ]
11. Carrillo-López N, Panizo S, Alonso-Montes C, Román-García P, Rodríguez I, Martínez-Salgado C, et al. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int. 2016;90:77-89. [ Links ]
12. Huang JC, Sakata T, Pfleger LL, Bencsik M, Halloran BP, Bikle DD, et al. PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res. 2004;19:235-244. [ Links ]
13. Coen G, Ballanti C, Mantella D Manni M, Lippi B, Pierantozzi A, et al. Bone turnover, osteopenia and vascular calcifications in hemodialysis patients. A histomorphometric and multislice CT study. Am J Nephrol. 2009;29:145-52. [ Links ]
14. Graciolli FG, Neves KR, dos Reis LM, Graciolli RG, Noronha IL, Moysés RM, et al. Phosphorus overload and PTH induce aortic expression of Runx2 in experimental uraemia. Nephrol Dial Transplant. 2009;24:1416-21. [ Links ]
15. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319-27. [ Links ]
16. Martínez-Arias L, Solache Berrocal G, Panizo García S, Carrillo López N, Avello Llano N, Quirós Caso C, et al. Efecto del sistema RANK/RANKL/OPG sobre la desmineralización ósea y la calcificación vascular en la enfermedad renal crónica. Rev Osteoporos Metab Miner. 2016;8(4):105-114. [ Links ]
17. Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260-8. [ Links ]
18. Orita Y, Yamamoto H, Kohno N, Sugihara M, Honda H, Kawamata S, et al. Role of osteoprotegerin in arterial calcification: development of new animal model. Arterioscler Thromb Vasc Biol. 2007;27:2058-64. [ Links ]
19. Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, López-Ongil S, et al. RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ Res. 2009;104:1041-8. [ Links ]
20. Osako MK, Nakagami H, Shimamura M, Koriyama H, Nakagami F, Shimizu H, et al. Cross-talk of receptor activator of nuclear factor-kappaB ligand signaling with renin-angiotensin system in vascular calcification. Arterioscler Trhromb Vasc Biol. 2013;33:1287-96. [ Links ]
21. Cejka D, Herberth J, Branscum AJ, Fardo DW, Monier-Faugere MC, Diarra D, et al. Sclerostin and Dickkopf-1 in renal osteodystrophy. Clin J Am Soc Nephrol. 2011;6:877-82. [ Links ]
22. Ferreira JC, Ferrari GO, Neves KR, Cavallari RT, Dominguez WV, Dos Reis LM, et al. Effects of dietary phosphate on adynamic bone disease in rats with chronic kidney disease--role of sclerostin?. PLoS One. 2013;8(11):e79721. [ Links ]
