










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La heparina, del griego “hepar” (hígado) fue descubierta en 19161. Este compuesto, es considerado un antitrombótico2,3que actúa por acción indirecta mediante la inhibición de varios factores de la cascada de coagulación4,5. Ejerce su mecanismo de acción anticoagulante, como catalizador, al potenciar la actividad inhibitoria de la antitrombina y otros inhibidores de proteasas de serina6. Es administrado en la fase aguda del tratamiento del tromboembolismo venoso7, en la prevención y el tratamiento de la trombosis6(venosa profunda y embolismo pulmonar), en intervenciones quirúrgicas7, en la hemodiálisis para prevenir la coagulación y otros procedimientos3.
El tratamiento del tromboembolismo venoso se realiza con la aplicación de heparina por vía subcutánea o mediante bolo intravenoso (con una dosis de carga) seguido de perfusión intravenosa contínua. Para el embolismo pulmonar se administra por vía intravenosa6.
Proveniente del uso de este medicamento se han presentado algunas complicaciones como trombocitopenia8, hematomas y hemorragia en el lugar de la cirugía9.
La heparina es un grupo heterogéneo denominado mucopolisacáridos sulfatados3,10con pesos moleculares entre 6000 y 20000 Da11. Se obtiene a partir de tejidos de mamíferos como la mucosa intestinal de los cerdos, bovinos u ovejas; y de los pulmones del ganado3,12.
En diferentes países se comercializa el medicamento con las marcas Lipo-Hepin®, Liquaemin® y Panheparin®13. En Cuba, los Laboratorios Liorad, de la Empresa Laboratorios AICA, como titular del Registro Médico Sanitario, elabora y distribuye una solución para inyección, para uso exclusivo de hospital, con el nombre heparina sódica 5.000 UI/mL.
Durante los estudios de estabilidad realizados a varios lotes, con la formulación inscrita, a partir de los seis meses en vida de estante se observó una disminución de valores de pH hacia el límite inferior de la especificación (5,5), identificándose como una inestabilidad química. En este trabajo se estableció como objetivo desarrollar una nueva formulación del producto heparina sódica 5.000 UI/mL,que mantenga los valores de pH en el rango especificado del producto (5,5-8,0)12a la vez que cumpla con los parámetros de potencia del Ingrediente Farmacéutico Activo (IFA) y el resto de requisitos para cualquier inyectable.
MATERIAL Y MÉTODOS
Evaluación retrospectiva del comportamiento del producto
En este trabajo, se efectuó un análisis documental de los registros correspondientes a los procesos tecnológicos de los lotes de heparina sódica (a partir de 2.015, 22 lotes). Además de la verificación de proveedores del IFA empleado en la manufactura de estos y los valores de pH final de las formulaciones.
Estudios de la formulación vigente
Se ejecutó un estudio de máxima estabilidad de pH durante 45 días, con la formulación vigente; en el que se elaboraron seis preparaciones con ajuste del pH en intervalos de 0,5 unidades dentro del rango de valores establecido por especificación (5,5 - 6,0 - 6,5 - 7,0 - 7,5 - 8,0).
Debido a que la estructura química de la heparina sódica puede ser afectada con la temperatura, se estudió la formulación mediante dos variantes14. La variante A, entre 15 °C y 25 °C (20 °C) y la B a 30 °C, comprobando además, el efecto de la temperatura en los valores de pH15.
La influencia de cada componente de la fórmula sobre el pH (5,5-8,0) se evaluó en otro estudio. Fueron preparadas cuatro soluciones, y cada ingrediente empleado estuvo en correspondencia con la cantidad declarada en el producto terminado: A- heparina sódica disuelta en agua, B- solución A con adición de cloruro de sodio, C- solución A con adición de clorobutanol y D- solución A agregándole metabisulfito de sodio. En todas se comprobó el pH final de la preparación y el comportamiento de este atributo en los intervalos de tiempo: 0, 24, 48, 72 y 96 horas. Un análisis extendido a 7, 14, 21, 28, 45 y 90 días complementó el estudio. Las muestras se almacenaron a temperaturas de 30 ± 2 °C y humedad relativa de 70 ± 5 %.
Análisis del IFA Heparina sódica
Los ensayos al IFA se verificaron según las especificaciones y límites de aceptación de la Farmacopea Británica (BP, por sus siglas en inglés) 2015. Los ensayos físico-químicos realizados fueron: características y solubilidad, identificación, apariencia de la solución, pérdida por desecación, metales pesados, impurezas proteicas y nucleotídicas, pH, contenido de sodio, cenizas sulfatadas y contenido de nitrógeno. Los ensayos microbiológicos estudiados fueron: esterilidad, endotoxinas bacterianas, carga microbiana y la valoración del IFA.
Elaboración de la formulación de heparina sódica 5.000 UI/mL
Etapa de preformulación
En el estudio de preformulación fueron consultadas varias presentaciones de diferentes fabricantes. Entre los componentes de uso común de estas formulaciones estuvieron los agentes conservantes como el alcohol bencílico2,16,17(concentración < 2 % v/v18) y los parabenos19,20(metilparabeno: 0,18 % w/v y propilparabeno: 0,02 % w/v18). Para garantizar la isotonicidad se utilizó el cloruro de sodio16(≤ 0,9 % w/v18) y como vehículo de uso general, el agua para inyección. De esta revisión fueron seleccionadas dos variantes a estudiar y una tercera (Tabla 1) en la que fueron empleados otros excipientes como el clorobutanol (preservo antimicrobiano en concentración < 0,5 % w/v18), el fosfato de sodio monobásico y el fosfato de sodio dibásico (2,23 % w/v18) como reguladores de pH y para obtener un producto terminado isoosmótico.
Tabla 1: Variantes de formulación.
| Componentes | Variantes | Función | Rango de uso | ||
|---|---|---|---|---|---|
| 116 | 219 | 3 | |||
| Heparina sódica | x | x | x | IFAa | (5 000 UI/mL) |
| Cloruro de sodio | x | x | Isotonizante | ≤ 0,9 % w/v | |
| Alcohol bencílico | x | Preservo | < 2 % v/v | ||
| Metilparabeno | x | Preservo | 0,18 % w/v | ||
| Propilparabeno | x | Preservo | 0,02 % w/v | ||
| Clorobutanol | x | Preservo | < 0,5 % w/v | ||
| Fosfato de sodio dibásico | x | Tampón | 2,23 % w/v | ||
| Fosfato de sodio monobásico | x | Tampón | 2,23 % w/v | ||
| Agua para inyección | x | x | x | Vehículo | c.s.pb. |
aIngrediente Farmacéutico Activo
bc.s.p. Cantidad suficiente para.
La concentración de agentes reguladores de pH a emplear en la variante tres fue calculada según se describe en el epígrafe “cálculos de pH y soluciones amortiguadoras”21de la Farmacopea de los Estados Unidos (USP, por sus siglas en inglés). Con la finalidad de garantizar un pH en el rango de especificación del producto terminado y un aporte a la isoosmoticidad de la solución, esta última fue comprobada comparando con una solución de cloruro de sodio 0,9 %.
Lotes a escala de Laboratorio
En la determinación de endotoxinas bacterianas fue aplicado el método de Lisado de Amebocitos del Limulus(LAL) en su variante cromogénica cinética. La curva estándar fue obtenida con el empleo de agua reactivo LAL como blanco a partir de soluciones de concentración 0,005; 0,05; 0,5; 5,0 y 50,0 UE/mL. La máxima dilución válida (MDV), fue estimada y determinada mediante el test de interferencias con varias diluciones del producto a las que se marcó con una concentración de endotoxinas de 4 λ y una serie de diluciones no marcadas de lotes en estudio (sin diluir, 1:10, 1:100 y 1:1000). La dilución de trabajo seleccionada fue determinada y validada.
El método de filtración por membrana fue el aplicado en el ensayo de esterilidad. Para definir el volumen de lavado a emplear fue necesario comprobar la actividad bactericida y fungicida del producto para lo cual se emplearon las cepas certificadas Bacillus subtilisATCC 6633, Staphylococcus aureusATCC 6538, Candida albicansATCC 10231 y Aspergillus nigerATCC 16404. En la valoración biológica se determinó la actividad anticoagulante de la heparina in vitro, mediante la comparación de su capacidad en condiciones dadas para retrasar la coagulación de plasma citratado recalcificado de ovejas, con la misma capacidad de una preparación de referencia de la heparina calibrada en unidades internacionales. La estandarización se efectuó con la evaluación de la linealidad, exactitud, precisión y especificidad.
Otros atributos de calidad (características organolépticas, identificación, acidez o alcalinidad, partículas visibles, partículas en inyectables y evaluación de la eficacia antimicrobiana) fueron evaluados según lo descrito en la BP 2015.
En los tres lotes de laboratorio se evaluaron los requisitos de calidad en el tiempo inicial luego de su elaboración, según farmacopea. La estabilidad en vida estante se valoró mediante el comportamiento de las características organolépticas, partículas visibles, pH y potencia biológica. La frecuencia planificada para el estudio fue: 0, 7, 14, 21, 28, 60, 90 y 120 días.
Escalado a lotes piloto
Para el escalado, se elaboraron tres lotes pilotos en la planta de producción que fueron sometidos a control de calidad recién elaborados y a estudios de estabilidad acelerados (6 meses) y vida de estante (24 meses). Los resultados positivos de estos estudios permitieron presentar el registro sanitario a la entidad regulatoria.
Lotes industriales
Una vez otorgado el Registro Sanitario de Medicamentos se realizaron las transferencias tecnológica y analítica a la planta de producción y laboratorios de control de la calidad, respetivamente. Se fabricaron tres lotes a escala industrial con los correspondientes controles de calidad y fue establecido un protocolo para realizar los estudios de estabilidad.
RESULTADOS
El cambio de proveedores del IFA (Hebei Changshan,China y Gland Pharma, India) no influyó en la preparación de la formulación con la que se elaboraron tres lotes.
Al evaluar los resultados del ensayo de acidez o alcalinidad se observó que, en el proceso de elaboración, el valor de pH final fue ajustado a 7,0.
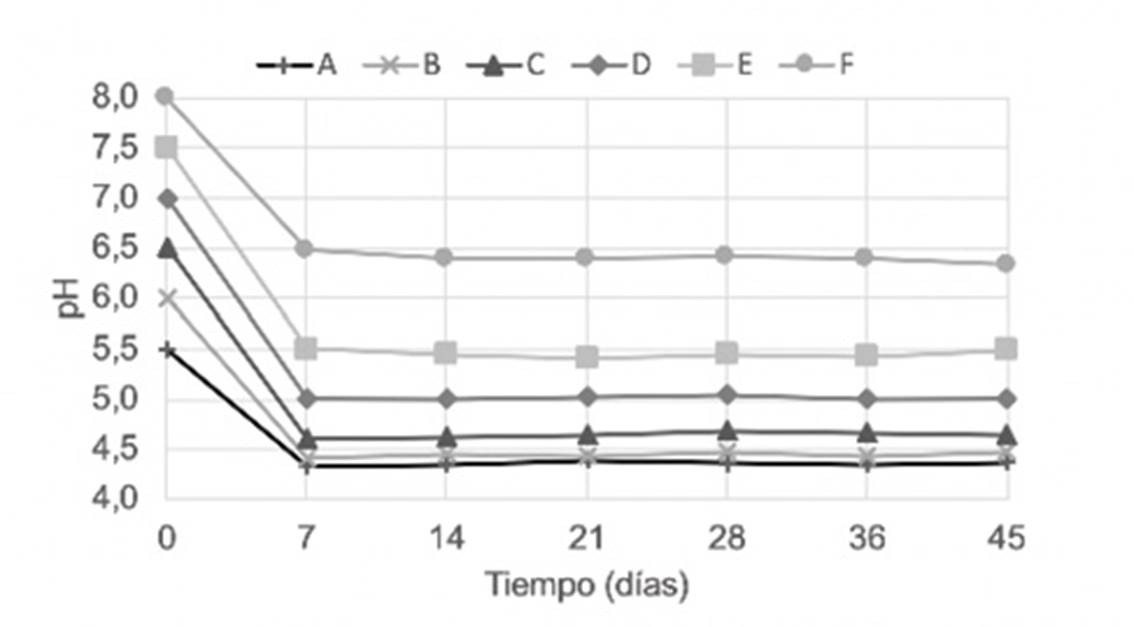
El estudio de máxima estabilidad de pH se muestra en la Figura 1.
En la Figura 2 se puede observar que la temperatura en la formulación no afectó el pH de la formulación.

Al evaluar la influencia de cada componente de la formulación sobre el pH se dedujo que el excipiente metabisulfito de sodio afectó este atributo de calidad.
En la Figura 3 se graficaron los valores de pH medidos en el tiempo, de las tres variantes de formulación ensayadas.

Los tres lotes, desarrollados en la etapa de laboratorio con la formulación seleccionada (variante tres), resultaron en soluciones incoloras, libres de turbidez, partículas visibles y sin sedimentación. Los otros resultados se muestran en la Tabla 2.
Tabla 2. Control de calidad de los lotes a escala de laboratorio.
| Ensayos / Lotes | 1 | 2 | 3 | |
|---|---|---|---|---|
| Acidez o alcalinidad (5,5-8,0) (uso de tampones) | 6,90 | 6,88 | 6,90 | |
| Volumen (mL) (No menor de 5,3 mL) | 5,4 | 5,3 | 5,4 | |
| Potencia Biológica | Potencia en valores relativos, 90-111% (Potencia en valores absolutos,UI/mL) | 100,6 % (5.032,1) | 99,8 % (4.991,7) | 101,1 % (5.052,7) |
| Límite fiduciales valores relativos (P= 0,95, 80-125%) (Límite fiducial valores absolutos, UI/mL) | 100,1 % -101,1 % (5.007,4-5.056,9) | 99,3 % -100,4 % (4.964,3-5.019,3) | 100,4 % -101,7 % (5.018,4-5.087,1) | |
| Conteo de partículas | ≥ 10 μm (≤ 6.000) | 3 | 3 | 3 |
| ≥ 25 μm(≤ 600) | 1 | 1 | 1 | |
| Endotoxinas bacterianas (No mayor que 10 UE/mL) | 0,3 | 0,4 | 0,3 | |
En cuanto a la estandarización de las técnicas analíticas, se definió y validó para la determinación de endotoxinas bacterianas como dilución de trabajo 1/100. El ensayo de esterilidad permitió corroborar que el producto en evaluación no presentó efecto bacteriostático ni fungistático, El ensayo biológico de valoración, resultó lineal, exacto, preciso y específico para su aplicación12.
El estudio de estabilidad vida estante reveló que las características organolépticas de los tres lotes de la variante tres a escala de laboratorio, así como la ausencia de partículas visibles, los resultados de la potencia biológica y el pH se mantuvieron dentro de los límites establecidos en el tiempo estudiado (Tablas 3 y (4).

Tabla 4. Resultado de potencia biológica de los lotes a escala de laboratorio.
| Parámetros | Lotes | Tiempo | |
|---|---|---|---|
| Inicial | Final | ||
| Potencia en valores relativos, 90-111% (Potencia en valores absolutos,UI/mL) Límite fiduciales valores relativos (P= 0,95, 80-125%) (Límite fiducial valores absolutos, UI/mL) | 1 | 100,6 (5.032,1) 100,1-101,1 (5.007,4-5.056,9) | 98,1 (4.906,7) 97,0-99,2 (4.851,9-4.962,2) |
| 2 | 99,8 (4.991,7) 99,3-100,4 (4.964,3-5.019,3) | 98,1 (4.906,0) 97,0-99,2 (4.851,9-4.962,2) | |
| 3 | 101,0 (5.052,7) 100,4-101,7 (5.018,4-5.087,1) | 100,0 (5.000,0) 99,3-100,7 (4.963,0-5.037,2) | |
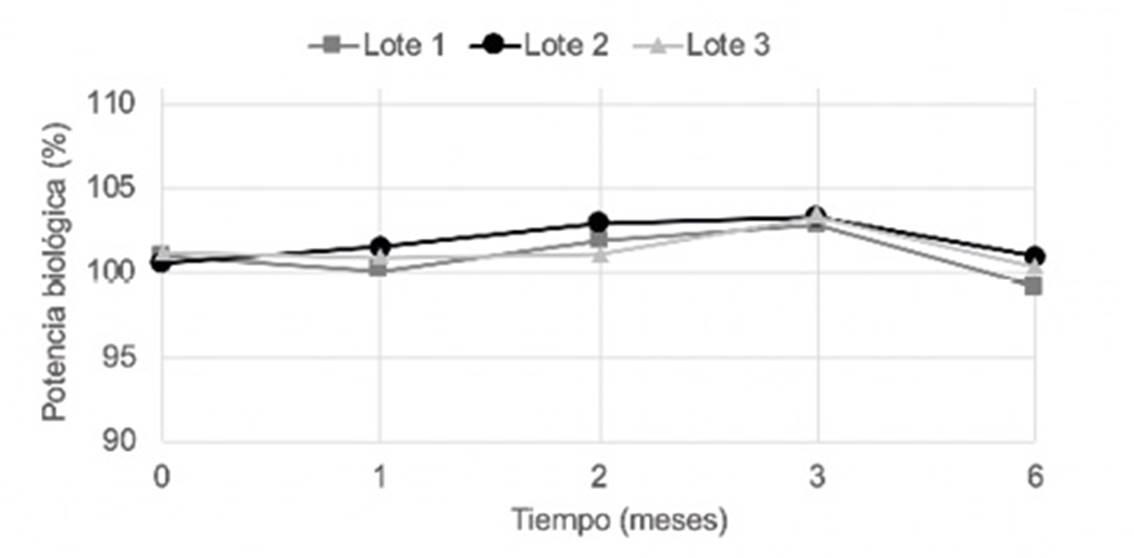
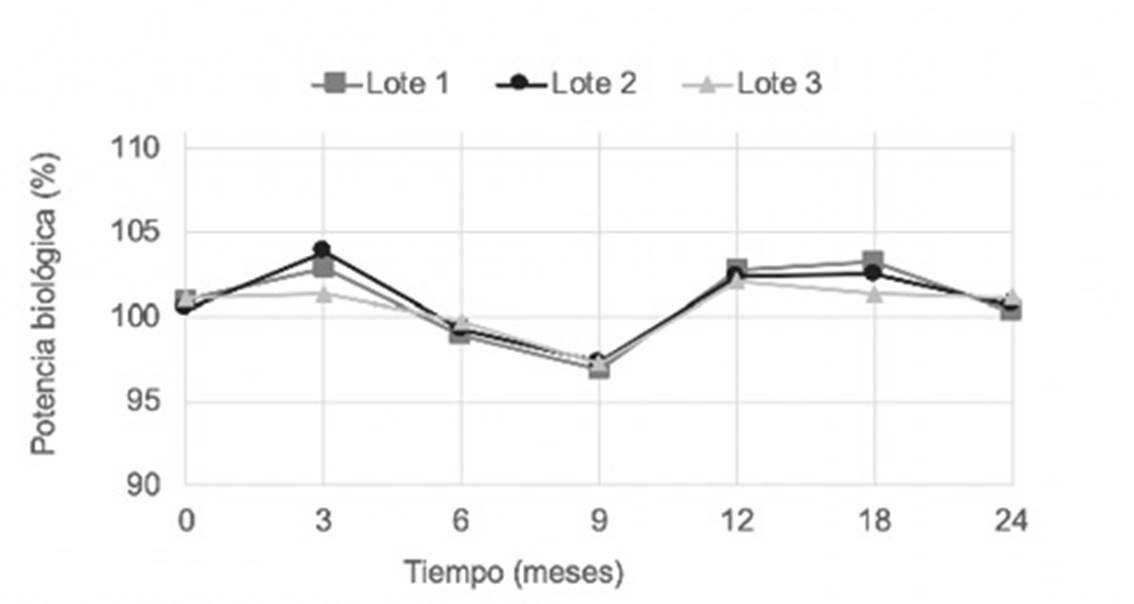
Los resultados obtenidos de potencia biológica en el estudio de estabilidad acelerada (6 meses) y vida estante (24 meses) se muestran en las Figuras 4 y 5.
DISCUSIÓN
La procedencia del IFA no afectó la estabilidad del pH, en todos los lotes evaluados, los resultados fueron similares,
En los 22 lotes de productos, fabricados de acuerdo a la formulación vigente, disminuyó el pH (pH= 5,6-6,8) con respecto al punto inicial, pero dentro de los límites especificados (5,5-8,0). Estos resultados sugirieron evaluar la formulación existente.
Para el estudio de pH que se aprecia en la Figura 1 los valores de este parámetro disminuyeron en el tiempo, independiente del valor de ajuste inicial.
Independiente de la temperatura (20 ó 30°C) empleada en las formulaciones estudiadas, se obtuvo un comportamiento similar en el tiempo para ambas temperaturas (Figura 2).
En la evaluación a la formula vigente, el metabisulfito de sodio, produjo una variación de 1,7 unidades de pH, durante el tiempo estudiado. Los otros componentes de la fórmula solo causaron una variación máxima de 0,7 unidades. Esto conllevó a reformular el producto y prescindir del metabisulfito de sodio, con ello se mantuvo la estabilidad del medicamento, especialmente su pH, sin presentarse inestabilidad química observable.
A los tres meses de estudio con las diferentes formulaciones, la variante uno tuvo desviación del pH en 1,2 unidades, la variante dos, en 0,4 unidades y la tres mostró cambios inferiores a 0,1 unidades de pH. Al evaluar la isotonicidad de esta última, a tres niveles de concentración de los agentes reguladores de pH, se aseguró la selección de una concentración que garantizó niveles de mOsmol en la formulación final equivalentes a una solución de cloruro de sodio 0,9%.
La variante tres fue escalada a nivel de laboratorio y los puntos críticos del proceso tecnológico fueron definidos (incorporación lenta de los búferes, adición de clorobutanol, y adición de heparina sódica, disolución mediante agitación constante de cada uno de los solutos durante 20 minutos). El proceso se llevó a cabo mediante procesamiento aséptico, con una filtración esterilizante al final.
Los resultados (Tabla 2) del control de calidad físico-químico y microbiológico de los lotes a escala de laboratorio, recién elaborados, cumplieron con las especificaciones de calidad establecidas; y demostraron la factibilidad de la tecnología propuesta para elaborar una formulación parenteral estéril en solución acuosa de este producto.
La dilución de trabajo para determinación de endotoxinas bacterianas fue escogida al no mostrar interferencias con el método de ensayo. Con los resultados obtenidos en el ensayo de esterilidad no fue necesario definir un volumen de lavado.
El estudio de estabilidad acelerada mostró valores de pH dentro de los límites establecidos (4 meses) con baja variación en los tres lotes (ΔpH < 0,1).
Por los resultados obtenidos a escala de laboratorio con la formulación tres, resultó factible escalar a lotes pilotos esta variante. El estudio de estabilidad, permitió establecer el periodo de validez y las condiciones de almacenamiento del producto en el envase propuesto para su comercialización. Los lotes recién elaborados, cumplieron con los criterios de calidad establecidos para el producto terminado.
Los valores de pH obtenidos durante el estudio de estabilidad, vida estante y acelerado con el empleo de fosfato de sodio monobásico/fosfato de sodio dibásico permitieron comprobar que en los tres lotes no se apreció disminución de los valores de pH, ni presencia de productos coloreados o precipitados, ni pérdida o disminución de la actividad anticoagulante. En este último parámetro, la variación fue inferior al 5 %, respecto al momento inicial.
Posterior a la inscripción de la variante tres, en la Autoridad reguladora de medicamentos nacional (Centro para el Control Estatal de Medicamentos, Equipos y Dispositivos Médicos), se llevó a nivel industrial la nueva formulación inyectable.
CONCLUSIONES
Los lotes fabricados a nivel industrial, recién elaborados cumplieron los controles de calidad para su liberación y comercialización. La factibilidad del desarrollo tecnológico del inyectable heparina sódica 5.000 UI/mL en solución con la formulación seleccionada (variante tres) quedó demostrada con el cumplimiento de las especificaciones de calidad. El producto terminado cumple con los valores de pH establecidos por especificación. La estabilidad de este último parámetro quedó resuelta con la nueva formulación. Se logró un inyectable con estabilidad física, química y microbiológica al lograr un producto trasparente, con la potencia correcta, estéril y apirogénico. Las condiciones de trabajo y preparación de los lotes garantizaron un estricto cumplimiento de las Buenas Prácticas de Fabricación establecidas para la producción de inyectables.

