










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales de Medicina Interna
Print version ISSN 0212-7199
An. Med. Interna (Madrid) vol.21 n.7 Jul. 2004
| Hepatitis autoinmune J. A. ORTS COSTA, A. ZÚÑIGA CABRERA1, I. ALARCÓN TORRES2 Área de Diagnóstico Biológico. Unidad de Autoinmunidad. 1Unidad de Biología
|
| AUTOIMMUNE HEPATITIS
| ||
| RESUMEN La hepatitis autoinmune es una inflamación hepatocelular que se presenta cuando el sistema inmunológico actúa frente a los hepatocitos y que se caracteriza por hallazgos histológicos (hepatitis de interfase con afectación periportal, infiltración de células plasmáticas y necrosis en sacabocados), bioquímicos (hipertransaminasemia e hipergammaglobulinemia) y autoinmunes (presencia de ciertos autoanticuerpos). Este trastorno es relativamente poco frecuente y suele afectar a mujeres de mediana edad. Al no existir un marcador patognomónico para su diagnóstico se requiere una exclusión cuidadosa de otras causas de enfermedad hepática conjuntamente con un patrón clínico y analítico compatible. El criterio de puntuación propuesto por el Grupo Internacional de la Hepatitis Autoinmune para su diagnóstico no es suficientemente específico como para definir la prognosis y el tratamiento. La hepatitis autoinmune se clasifica, según los autoanticuerpos presentes, en subtipos 1 y 2, aunque esta clasificación no muestra repercusiones clínicas importantes. El anteriormente conocido como subtipo 3 no difiere clinicamente del 1 en forma significativa y por tanto debe incluirse dentro de este último grupo. Todavía permanecen por dilucidar la etiología y las bases moleculares de esta enfermedad, originada probablemente por la interacción de diversos factores como la predisposición genética (haplotipos HLA-DR3 y DR4), pérdida de tolerancia inmunológica, formación de neoantígenos por factores desencadenantes como virus o fármacos, y por mimetismo molecular. La terapia inmunosupresora (corticosteroides, azatioprina) ofrece excelentes resultados. Nuestro objetivo es revisar esta enfermedad bajo diferentes puntos de vista, considerando: los aspectos clínicos, histopatológicos, etiológicos, genéticos, bioquímicos, autoinmunes, de tratamiento y pronóstico. PALABRAS CLAVE: Hepatitis autoinmune. Anticuerpos antinucleares (ANA). Anti-LKM. Anticuerpos anti-músculo liso (SMA). | ABSTRACT Autoimmune hepatitis (AIH) is a hepatocellular inflammation that is characterised by a wide range of histolopatholgic (periportal interface hepatitis with plasma cell infiltration and piecemeal necrosis), biochemical (hypertransaminasemia, hypergammaglobulinaemia) and autoimmune (several autoantibodies presence) features. This relatively rare disorder frequently affects middle-aged women. There is no pathognomonic marker for AIH diagnosis, therefore it requires a careful rule out of other causes of liver disease together with the detection of a suggestive pattern of clinical and laboratory abnormalities. Scoring system for AIH diagnosis proposed by International Autoimmune Hepatitis Group has been used as a tool in clinical practice but is not sufficiently exclusive in terms of defining prognosis or treatment. AIH has been classified in two subtypes according to autoantibodies detected: 1 and 2, but this classification results in poor clinical implications. Previously known as subtype 3 is at the present included in subtype 1 because no clinical significant differences has been found between them. Aetiology, and molecular mechanisms still remain to be elucitaded in this disease, although viruses, drugs and molecular mimicry act presumably as a trigger in genetically predisposed patients (associated with HLA-DR3 and DR4 haplotypes). On the other hand, immunosuppresive therapy (corticosteroid or azathioprine) generally offers favourable response. Our aim is to review this disease from different points of view, considering: clinical, histopathological, etiologic, genetic, biochemical, autoimmune, treatment and prognosis features. KEY WORDS: Autoimmune hepatitis. Antinuclear antibodies (ANA). Liver-kidney microsomal autoantibodies (anti-LKM). Smooth muscle autoantibodies (SMA). |
Orts Costa JA, Zúñiga Cabrera A, Alarcón Torres I. Hepatitis autoinmune. An Med Interna (Madrid) 2004; 21: 340-354.
Trabajo aceptado: 5 de marzo de 2004
Correspondencia: Juan Antonio Orts Costa. Hospital de la Ribera. Ctra. Alzira-Corbera, km. 1. 46600 Alzira. Valencia. e-mail: jaorts@hospital-ribera.com
INTRODUCCIÓN
Conocida inicialmente como "hepatitis crónica viral", se considera que fue descrita con rigor por primera vez en 1950 por Waldeström, al observar en Suecia un tipo de hepatitis persistente que afectaba principalmente a mujeres jovenes y se asociaba con infiltración hepática de células plasmáticas, hipergammaglobulinemia, amenorrea y manifestaciones dermatológicas.
En 1955, Joske describió la presencia del fenómeno LE (anticuerpos antinucleares, ANA) en hepatitis crónicas activas y en 1956 MacKay la denominó (erróneamente, pues no está relacionada con el lupus) como hepatitis lupoide. También fue denominada como: cirrosis con síndrome adrenogenital y hepatitis plasmocelular. Posteriormente, en 1966, Whittingham observó la asociación con anticuerpos anti-músculo liso (SMA) y denominó la enfermedad como hepatitis crónica activa autoinmune, estableciéndola como una entidad diferenciada.
Actualmente, la HAI se considera como una enfermedad necro-inflamatoria del hígado, infrecuente, usualmente crónica y progresiva, de etiología no suficientemente conocida que se caracteriza por la presencia de alteraciones inmunológicas entre las que se encuentran la hipergammaglobulinemia y la presencia de autoanticuerpos, con una buena respuesta al tratamiento con inmunosupresores y mayor prevalencia en mujeres. No existe un marcador patognomónico de esta enfermedad. La enfermedad quedó definida en 1992 por el Grupo Internacional de Hepatitis Autoinmune (GIHA) siguiendo un sistema de puntuación que fue revisado posteriormente (1-2) (Tabla I). Basándose en diferentes patrones autoinmunitarios puede clasificarse dos subtipos: 1 y 2.

Los principales problemas que se encuentran para mantener un concepto unitario sobre las hepatitis autoinmunes son: la ausencia de rasgos clínicos y analíticos patognomónicos, dificultad para la identificación de los autoanticuerpos y autoantígenos implicados, y la compresión todavía parcial de los mecanismos inmunológicos responsables.
Se presenta con mayor prevalencia en individuos que muestran alelos HLA de clase II, haplotipos HLA DR3 y DR4, en mujeres y en aquellos portadores del alelo nulo del C4. Por lo general, la enfermedad es más severa en las pacientes más jovenes.
Debe sospecharse una hepatitis autoinmune cuando la afectación hepática -objetivable por la alteración de pruebas analíticas- perdura durante más de seis meses y se han descartado otras etiologías: víricas, metabólicas o genéticas. De todas ellas las que inducen a diagnóstico diferencial con más frecuencia son la hepatitis por VHC y la enfermedad de Wilson.
Las hipótesis actuales sobre la etiopatogenia de la HAI proponen como factores desencadenantes a agentes víricos hepatotropos, agentes ambientales y productos químicos o fármacos que, sobre huéspedes genéticamente predispuestos, inducirían la pérdida de tolerancia a los antígenos hepatocelulares que dispararía un ataque autoinmune.
El tratamiento, ya clásico y altamente eficaz, se basa en monoterapia corticoesteroidea o en la combinación de esteroides y azatioprina y suele ser de por vida ya que la remisión a largo plazo sólo se alcanza en un 17% de los pacientes. Si no es tratada, la HAI ofrece una pobre prognosis con desarrollo de insuficiencia hepática y cirrosis.
ETIOLOGÍA
Actualmente se desconoce la etiología de la enfermedad, sospechándose que en indivíduos genéticamente predispuestos algún agente ambiental activa el sistema inmune contra antígenos hepáticos, provocando una inflamación progresiva crónica. El mimetismo molecular con proteínas virales o con otros autoantígenos podría tener un papel clave en el desarrollo de la HAI.
La HAI se ha asociado con diferentes infecciones virales: herpesvirus, virus del sarampión, virus de la hepatitis A y B (3-7). También el virus de la hepatitis C (VHC) se ha relacionado con el fenómeno, pero los estudios realizados con análisis de PCR sólo han detectado genoma viral en una minoría de los pacientes (8). Igualmente, se ha descrito que el citocromo CYP450 2D6, uno de los autoantígenos reconocidos por los anticuerpos anti-LKM-1 presentes en la HAI tipo II, comparte similaridades antigénicas con el VHC (GOR-47) y el HSV tipo 1 (9). Fármacos como la minociclina, isoniazida, a-metildopa, hidralazina y nitrofurantoina, así como diversas toxinas y aditivos alimentarios también podrían desencadenar la HAI (10,11). En mujeres con hepatitis C crónica (HCC) y susceptibilidad genética, una HAI latente podría desarrollarse por los efectos inmunoestimuladores de la terapia con interferón-α (IFN-α) (12). La inducción o exacerbación de enfermedades autoinmunes hepáticas o extra-hepáticas por esta terapia se conoce desde hace tiempo y podría estar relacionada con la citotoxicidad celular dependiente o por la expresión aberrante de antígenos HLA con la consiguiente citotoxicidad mediada por linfocitos T (13).
ETIOPATOGENIA
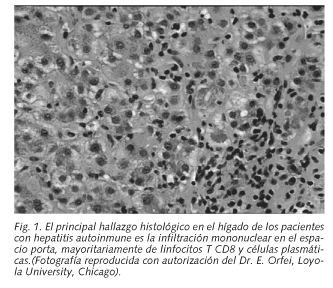
El principal hallazgo histológico en el hígado de los pacientes con HAI, es la infiltración mononuclear en los zonas de lesión, mayoritariamente de linfocitos T CD8 en los tractos portales inflamados (Fig. 1). Esto sugiere que tienen un papel patogénico en las áreas de daño o necrosis hepática. Además, hay numerosos datos que indican que moléculas clave de la interacción entre la célula efectora y la diana, con el subsiguiente proceso citolítico, tienen una expresión diferente en el hígado de los pacientes de HAI, lo que implica un claro origen inmunorreactivo de la enfermedad.
MOLÉCULAS HLA CLASE I Y CLASE II
Las moléculas HLA I se ha demostrado que se expresan en los hepatocitos tanto en hígados normales como en dañados por diferentes enfermedades. Las moléculas HLA clase II se expresan de un modo aberrante en los hepatocitos de los pacientes con HAI, y potencialmente podrían contribuir a la patogénesis de la enfermedad (14).
AUTOANTÍGENOS
La expresión hepatocelular de moléculas HLA I y II no es suficiente para iniciar una respuesta inmune; es necesaria la expresión de un autoantígeno en la membrana de los hepatocitos para que sean reconocidos por las células T. Se han descrito hasta la fecha dos posibles antígenos de superficie: a) CYP450 2D6, reconocido por los anti-LKM-1 (15), y b) receptor de asialoglucoproteínas unido a membrana, específico de los hepatocitos, que se expresa preferentemente en las zonas periportales y que está asociado con títulos altos de anticuerpos circulantes en la HAI (16).
También se ha demostrado la participación de anticuerpos frente al antígeno soluble hepático (anti-SLA) y frente a un antígeno citoplasmático compartido por el hígado y el páncreas (anti-LP). Se ha visto que ambos anticuerpos son el mismo y que éste es muy específico de la HAI; teniendo como diana autoantigénica la proteína asociada a t-ARN UGA-supresora (17,18). Otro anticuerpo estudiado ha sido el anti-LC1 que reacciona con antígenos citosólicos hepatoespecíficos: la argininosuccinato liasa (19) y la ciclodeaminasa formiminotransferasa (20).
MOLÉCULAS DE ADHESIÓN CELULAR
En condiciones basales, las moléculas de adhesión responsables de la interacción inicial antígeno-independiente entre las células T citotóxicas y las células dianas se expresan a niveles muy bajos. Sin embargo, los mediadores de la inflamación, incluyendo a las citoquinas, causan una fuerte inducción de estas moléculas, lo que se ha demostrado claramente en el caso de la HAI. ICAM-1/CD54 se induce en los pacientes con HAI especialmente en áreas de inflamación periportal (21). ICAM-1 es el contrarreceptor de la célula diana del antígeno de función leucocitaria (LFA-1, CD11c/CD18). La interacción entre las dos moléculas es crucial en el desencadenamiento de la respuesta inmune. De un modo similar, LFA-3/CD58 (contrarreceptor en la célula diana de LFA-2/CD2 presente en las células T) se expresa en la membrana de los hepatocitos de los pacientes con HAI y no en la de los sanos (22).
La sobrexpresión de antígenos de activación linfocitaria y de las moléculas de adhesión celular en la HAI, es una evidencia clara de que los mecanismos inmunes mediados por células T están implicados en la patogénesis de la HAI.
CITOQUINAS
No se conoce cuál es el repertorio de citoquinas implicadas en la HAI. Una excesiva o desequilibrada producción de citoquinas podría ser un mecanismo por el que se produzca una estimulación anormal de los linfocitos, incluyendo a las células autorreactivas. Varias citoquinas están implicadas en la patogénesis de enfermedades hepáticas caracterizadas por la inflamación y la fibrosis (23). Se ha descrito en la HAI una elevada producción de IL-4 por los linfocitos T infiltrados (24). También existen evidencias de otras citoquinas implicadas como el TGF-ß1 o CIIP-10; sin embargo estos fenómenos no son específicos de la HAI y se ha observado en otras patologías hepáticas (25,26).
NUEVOS FACTORES PATOGÉNICOS
En pacientes con HAI se ha observado un incremento en la expresión de enzimas implicadas con el daño oxidativo a estructuras celulares por lo que también podría estar relacionado con la patogénesis de la HAI (27).
Otro hallazgo interesante es la formación de nueva vasculatura en las áreas de inflamación portal de los pacientes de HAI, lo cual indica que la angiogénesis como respuesta adaptativa al daño hepático también se encuentra presente en esta enfermedad (9).
BASES GENÉTICAS DE LA HEPATITIS AUTOINMUNE
La presentación del autoantígeno, la activación del sistema inmune, la diferenciación de la célula efectora y el daño hepático están influenciados por factores genéticos, la caracterización de estos factores ayudaría a un mejor diagnóstico y al desarrollo de algoritmos más precisos (28,29).
PREDISPOSICIÓN FEMENINA A LA HAI
La mujer tiene una mayor predisposición a padecer HAI, así como otras enfermedades autoinmunes, pero se desconoce la causa de este hecho, especulándose sobre si los mecanismos inmunomoduladores están controlados por genes ligados al sexo u hormonas sexuales que actuarían sobre el sistema inmunitario y/o los alelos de susceptibilidad (30). Un gen inmunomodulador en el cromosoma X podría ser la causa, por el efecto doble dosis que se daría en la mujer, de la mayor inmunorreactividad. Otra posibilidad sería que los estrógenos y otras hormonas sexuales tuvieran influencia en el vigor de la respuesta facilitando, mediante algún mecanismo, el procesado y reconocimiento del antígeno.
HAPLOTIPOS DE RIESGO EN LA HAI
En la HAI tipo 1 el alelo HLA DRB1*0301; que codifica para el antígeno HLA DR3 y frecuente en la raza caucásica es considerado como factor de susceptibilidad. Un factor de riesgo secundario e independiente es el DRB1*0401, que codifica el antígeno HLA DR4. El 84% de los pacientes del Reino Unido y de EE.UU. son portadores de uno o de los dos alelos (31). Sin embargo, en Japón el principal alelo de susceptibilidad es el DRB1*0405; en Argentina entre los adultos es el DRB1*0405 y entre los niños el DRB1*1301; en los mestizos mejicanos el DRB1*0404; y en Brasil los principales alelos de susceptibilidad encontrados han sido DRB1*13 y DRB1*03 (29).
En la HAI tipo 2 los alelos de susceptibilidad encontrados son distintos a los de la HAI tipo 1. En Brasil y Alemania, el DRB1*07 es el principal factor de riesgo (32). Otros estudios apuntan hacia el HLA B14, DR3 y C4A-QO como contribuyentes a una mayor susceptibilidad en el portador. Estas asociaciones implican que el antígeno diana en la HAI tipo 2 y los factores genéticos de la enfermedad, son distintos a la HAI tipo 1. La identificación del CYP450 2D6 como un autoantígeno específico de la HAI tipo 2 apoya la idea anterior.
La multiplicidad de los alelos de riesgo dependientes de la raza en la HAI sugieren que alelos diferentes codifican uno o mas determinantes que son criticos en la expresión autoinmune, o que estos alelos son promotores independientes seleccionados por desencadenantes etiológicos especificos de la enfermedad (33).
ALELOS MHC CLASE II COMO DETERMINANTES DE SUSCEPTIBILIDAD Y RESISTENCIA
Las moléculas MHC clase II son glucoproteínas heterodiméricas con una cadena invariable DRα y una cadena polimórfica DRß, siendo una región de esta ultima (HVR3) la implicada en la interacción con el receptor de las células T. Las moléculas MHC clase II afectan a la respuesta inmune frente a antigenos extraños y propios. Actualmente se cree que en la herencia de alelos HLA clase II específicos, estaría la base de la susceptibilidad a la enfermedad debida a MHC. Los alelos HLA DRB1*0301 y DRB1*0401 relacionados con la HAI tipo I codifican en la región HVR3 del polipéptido DRß el mismo motivo de aminoácidos LLEQKR, siendo el aminoácido K (lisina) el residuo crítico. Este motivo se encuentra en el 94% de los pacientes de HAI tipo 1 de raza caucásica; en el caso de los alelos DRB1*0404 y DRB1*0405 asociados con la HAI tipo 1 en Japón y Méjico, el motivo es LLEQRR, donde la lisina (K) está sustituida por una arginina (R). Ambos aminoácidos están cargados positivamente y su cambio afecta mínimamente, por lo que el riesgo de padecer HAI tipo 1 sería similar en los pacientes portadores de estos alelos con independencia de sus diferencias étnicas.
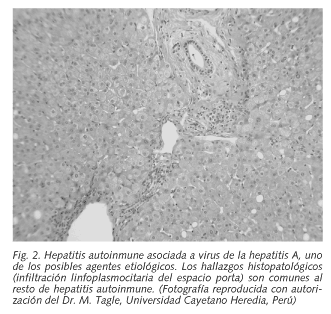
Frente a esta hipótesis del motivo compartido que sería la responsable de la susceptibilidad a la HAI tipo 1, los estudios realizados en Sudamérica son discrepantes lo que podría explicarse por ser distintos las causas detonantes de la enfermedad (entre ellas la infección por el virus de la hepatitis A) (5, 34) (Fig. 2).
HIPÓTESIS DEL PROMOTOR AUTOINMUNE
Esta hipótesis propone que la susceptibilidad y la expresión clínica de una enfermedad autoinmune son el reflejo de un efecto compuesto entre unos factores primarios de riesgo y/o promotores autoinmunes no específicos. En algunas circunstancias, múltiples promotores podrían facilitar la aparición o afectar a la evolución de la enfermedad en ausencia/presencia de factores de riesgo primarios.
Los polimorfismos en el promotor del gen factor de necrosis tumoral a (TNF-A) y del gen del antígeno 4 del linfocito T citotóxico (CTLA-4) en pacientes de HAI tipo 1 apoyan esta idea.
El polimorfismo -308 del promotor del gen TNFA*2 es muy frecuente en los pacientes de raza caucásica afectos de HAI tipo 1, este polimorfismo induce la sobrexpresión de un modo constitutivo del TNF-α, lo que alteraría el entramado de citoquinas que regulan la respuesta de la célula efectora T CD4. Los pacientes portadores presentan la enfermedad a una edad más temprana, fallan más con el tratamiento y progresan a cirrosis con mayor frecuencia (35).
De un modo similar, también se ha descrito otro polimorfismo (G) del gen CTLA-4 presente con mayor frecuencia en los pacientes de HAI tipo 1, y que se asocia con unos mayores niveles séricos de aspartato aminotransferasa y de anticuerpos microsomales tiroideos. Los individuos homocigotos G/G presentan parámetros bioquímicos sugerentes de daño hepatocelular más severo y con mayor inmunorreactividad (36).
Estos polimorfismos actuarían de un modo sinérgico con los alelos HLA que originan susceptibilidad. Podrían existir, de este modo, múltiples promotores no específicos de autoinmunidad de los que no se conoce prácticamente nada, sólo hay publicados trabajos sobre el alelo nulo C4 del complemento que se ha demostrado que actúa de un modo sinérgico con el alelo HLA DRB1*0301.
CRITERIOS DIAGNÓSTICOS Y CLASIFICACIÓN
HALLAZGOS HISTOPATOLÓGICOS
La biopsia hepática continúa siendo esencial en el diagnóstico y tratamiento de la HAI. La enfermedad se manifiesta como una hepatitis de interfase generalmente asociada a actividad lobulillar, amplias áreas de colapso reticulínico hipocelular, transformación microacinar de los hepatocitos (en roseta), infiltración periportal de células inmunitarias (linfocitos T y B, plasmocitos, macrófagos) y necrosis hepato-celular periportal (piecemeal, sacabodados) a veces con puentes (Fig. 1). Los hepatocitos en roseta se muestran hinchados, agrupados y separados del resto de hepatocitos dañados por la inflamación y el estroma colapsado. Estas rosetas se suelen hallar conjuntamente con infiltración de células plasmáticas. Clásicamente se ha pensado que la presencia de células plasmáticas es el hallazgo clave de la enfermedad, sin embargo en un 34% de los pacientes su presencia es escasa o ausente (37). Se excluyen hallazgos de lesiones biliares, granulomas bien definidos, u otros cambios prominentes que sugieran una etiología diferente. Sin embargo, estos hallazgos no son suficientemente específicos y pueden ser encontrados en todas las hepatitis crónicas (38,39).
En el curso de la enfermedad, la necrosis se extiende progresivamente, según la severidad de la enfermedad, hacia áreas centrales lobulillares. La necrosis y regeneración de los hepatocitos en la HAI muestran predominio periportal en contraste con el patrón focal observado en la hepatitis vírica. La fibrosis sigue un curso de ampliación portal y formación septal como en otras formas de hepatitis crónica. La apoptosis, observada en las principales hepatopatías crónicas, es menos frecuente en la HAI.
Pueden desarrollarse folículos linfoides genuinos, aunque son menos usuales que, por ejemplo, en la HCC.
En un tercio de los casos, los ductos biliares pueden estar afectados por un proceso inflamatorio y mostrar cambios degenerativos que no son debidos a colangitis auténtica. La diferencia esencial con la Cirrosis Biliar Primaria (CBP) radica en que en esta última existe afectación de la membrana basal ductal.
HALLAZGOS BIOQUÍMICOS
El patrón bioquímico sérico es el típico de una hepatitis, con cifras elevadas de aminotransferasas y bilirrubina, siendo la fosfatasa alcalina normal o ligeramente elevadas. El grado de elevación de las aminotransferasas no es predictivo del daño o la extensión de la fibrosis (37). La gammaglutamiltransferasa puede estar incrementada pero su significación es incierta. Es característica la hipergammaglobulinemia, por aumento de la IgG. La IgM puede estar moderadamente elevada pero la IgA es normal. La α1-antitripsina, ceruloplasmina y cobre en suero son generalmente normales aunque pueden variar ampliamente según individuos. Es importante remarcar que cifras bajas de aminotransferasas, bilirrubina o IgG no indican una afectación leve o excluyen necesariamente una HAI. Los pacientes afectos de HAI tienen a menudo niveles persistentemente bajos de C4 (40).
AUTOANTICUERPOS EN LA HAI
Se han asociado diferentes autoanticuerpos dirigidos a diversos autoantígenos en la HAI (la mayoría frente a epítopos correspondientes a moléculas intracelulares) que han sido intensamente estudiados y han servido de base para la clasificación de la HAI en dos grupos serológicos. De esta clasificación trascienden pocas consecuencias clínicas, pero los perfiles serológicos indican que la HAI es un grupo heterogéneo y que distintas causas pueden provocar una disminución de la tolerancia hacia diferentes autoantígenos. No hay ningún autoanticuerpo absolutamente específico de la HAI, pero su caracterización así como la de sus dianas antigénicas (Tabla II), ha conducido a la definición de cierta especificidad clínica (Fig. 3).


AUTOANTICUERPOS EN LA HAI TIPO 1
ANA y SMA
Los ANA y SMA son los marcadores característicos de la HAI tipo 1. Ambos autoanticuerpos se detectan conjuntamente en un 54% de los pacientes de etnia caucásica afectos de HAI, mientras que los ANA en solitario se encuentran en un 15% y los SMA en un 35%. Es decir, entre un 70-80% de los pacientes con HAI presentan títulos significativos de ANA, SMA o ambos. Sin embargo, ni la presencia de estos autoanticuerpos ni su título tienen valor patogomónico o pronóstico sobre la enfermedad. La inmunosupresión durante el tratamiento puede hacer desaparecer los autoanticuerpos (37,41).
Las dianas nucleares de los ANA, pese a las intensas investigaciones realizadas, todavía no están perfectamente definidas. Los ANA muestran reactividad frente a componentes funcionales o estructurales nucleares e incluso de membrana. Los antígenos diana son heterogénos y no completamente definidos: DNA (simple o doble cadena), tRNA, SSA-Ro, RNPs, lamininas A y C, ciclina A, e histonas. El procedimiento más frecuente para la determinación de los ANA es la inmunofluorescencia indirecta (IFI), siendo los patrones más frecuentes los homogéneos y moteados, y los títulos elevados (1/≥ 160) (37,42).
Los SMA son los anticuerpos más frecuentemente presentes en la HAI (hasta en un 87% de los pacientes) se dirigen contra las proteínas del citoesqueleto como la actina, troponina y tropomiosina. Se determinan, usualmente, por IFI en estómago de rata. Los títulos superiores a 1/80 de clase IgG son frecuentes en las HAI generalmente asociados a ANA, aunque no son específicos de la HAI y pueden encontrarse a títulos bajos (1/≤ 80) en otras enfermedades hepáticas, enfermedades infecciosas y trastornos reumáticos (42). Dentro de los SMA, los anti-F-actina son los predominantes y más específicos aunque pueden encontrarse en afectos de CBP a títulos bajos. La determinación de anti-F-actina no se realiza rutinariamente en los laboratorios, sin embargo títulos altos (1/>320) de SMA comúnmente reflejan indirectamente la presencia de anti-F-actina (43).
Anticuerpos anti-citoplasmáticos de patrón perinuclear (p-ANCAs)
Aunque se asocian generalmente a colangitis esclerosante primaria (CEP), se ha descrito su presencia a títulos altos en más del 65% de pacientes con HAI (44). Esta determinación es útil en la diagnosis de HAI tipo 1 seronegativa a ANA/SMA/LKM (un 20%), en el diagnóstico diferencial con hepatitis víricas positivas a ANA/SMA /LKM, y también para distinguir las HAI genuinas de los síndromes de solapamiento como la CBP (41). Se han relacionado con una mayor severidad y recurrencia de la enfermedad (45).
Anticuerpos contra los antígenos hepáticos solubles (anti-SLA) o frente al antígeno hígado-páncreas (anti-LP)
Estos anticuerpos, específicos de la HAI, se presentan en un 10% de los pacientes con ANA/SMA y en un 30% de pacientes de los que carecen de estos autoanticuerpos. Van dirigidos contra determinantes antigénicos (principalmente citoqueratina 8 y 18 del citoplasma de los hepatocitos), fueron en 1994 motivo de subclasificación de la HAI como HAI tipo 3. Sin embargo, recientes hallazgos genéticos, serológicos y clínicos han evidenciado que su presencia no justifica este nuevo subtipo y actualmente se incluye dentro de la HAI tipo 1 (41).
AUTOANTICUERPOS EN LA HAI TIPO 2
Anticuerpos contra los microsomas hepato-renales (anti-LKM)
Se han identificado tres tipos de anti-LKM. El característico para el diagnóstico de la HAI tipo 2 es el anti-LKM-1, ya que no ha sido descrito en ninguna otra enfermedad autoinmune. Se observa en un 3-6% de los pacientes con HAI. El principal autoantígeno identificado es el CYP450 2D6 y al menos cuatro epítopos diferentes han sido definidos.
Los anti-LKM-2 se han encontrado en algunos casos de hepatitis inducida fármacos (ácido tienílico, halotano, dihidralazina y agentes anticonvulsionantes) pero nunca, en afectos de HAI, se ha descrito como autoantígeno el CYP450 2C9.
Los anti-LKM-3 dirigidos contra el autoantígeno familia 1 de las UDP-glucuronosialtransferasas (1-UGTs) fueron descritos inicialmente en un 13% de los pacientes afectos de hepatitis D, sin embargo recientemente se han observado en algunos pacientes con infección por VHC, tratándose, probablemente y al igual que algunos LKM-1 observados en HCC, de epifenómenos autoinmunitarios dentro de esta enfermedad.
Anticuerpos frente a la proteína citosólica hepática tipo 1 (LC1)
Son detectados en aproximadamente el 30% de los pacientes con HAI tipo 2, son raros en pacientes mayores de 40 años. La mayoría (aproximadamente un 70%) se asocian con la presencia de anti-LKM-1. También se han encontrado en HCC y CEP. En aproximadamente el 10% de las HAI se trata del único marcador serológico detectado. Por IFI muestra fluorescencia citoplasmática de los hepatocitos periportales, también pueden determinarse por técnicas de inmuno-dots al igual que los SLA. Su presencia parece correlacionar con la actividad de la enfermedad (37).
AUTOANTICUERPOS COMUNES A LA HAI TIPO 1 Y 2
Anticuerpos frente al receptor de asialoglucoproteína (anti-ASGP-R)
El receptor de asialoglucoproteína (ASGP-R) se trata de un receptor hepático de membrana que se encuentra principalmente en los hepatocitos periportales, donde la necrosis en sacabocados se manifiesta como marcadora de la actividad inflamatoria severa. Su principal función consiste en la internalización de asialoglucoproteínas mediante la unión de la galactosa terminal y se especula sobre su participación en los mecanismos patogénicos de la HAI.
Los anti-ASGP-R se consideran marcadores universales de la HAI puesto que son positivos en ambos tipos, se presentan en hasta un 88% de los pacientes, correlacionan con la actividad clínica y disminuyen como respuesta a la inmunosupresión. Sin embargo, la determinación requiere ASGP-R purificado que todavía no es fácilmente disponible. Se ha descrito una HAI "criptogénica" caracterizada por la presencia de anti-ASGP-R, hipergammaglobulinemia y buena respuesta al tratamiento inmunosupresor. Por otra parte no son suficientemente específicos, ya que también se pueden encontrar en en otras afecciones hepáticas: hepatitis B crónica, hepatitis alcohólica, y CBP. Su presencia parece correlacionar con la actividad histológica y el tratamiento inmunosupresor (37).
CRITERIO DIAGNÓSTICO DEL GRUPO INTERNACIONAL SOBRE LA HEPATITIS AUTOINMUNE (GIHA)
En 1992 el GIHA estableció el diagnóstico de la HAI basándose en un sistema de puntuación sobre características clínicas, anatomopatológicas, bioquímicas y serológicas (Tabla I). Igualmente, sustituyó la denominación de hepatitis crónica autoinmune por la de hepatitis autoinmune. Los criterios fueron revisados y actualizados en 1999 (2). Este sistema de puntuación, descrito inicialmente como una ayuda en la selección de pacientes homogéneos para propuestas de investigación, ha probado su utilidad en la práctica clínica diaria, especialmente para pacientes atípicos o en algunos casos de solapamiento. Ha sido validado en pacientes en EEUU, Europa y Asia. Su sensibilidad para el diagnóstico de la HAI "cierta" o " probable" oscila entre el 97 y el 100% y su especificidad para excluir una HAI en pacientes con hepatitis C crónica varía entre el 66% y el 92%. Sin embargo se ha descrito que su capacidad para discernir la CEP y otros síndromes colestásicos de la HAI no es buena. Es útil para el diagnóstico inicial y como guía para instituir la terapéutica adecuada, pero sin embargo, no provee información adicional sobre la severidad de la enfermedad o su prognosis (37).
Para el diagnóstico de HAI "cierta" se requiere ausencia de marcadores de replicación viral (incluidos CMV y VEB), ausencia de exposición a hemoderivados, alcohol y fármacos hepatotóxicos, positividad para anticuerpos antinucleares (ANA), antimúsculo liso (SMA) o antimicrosomas higado-riñon (LKM-1) a títulos superiores o iguales a 1/80, niveles de gammaglobulinas superior a 1,5 el nivel de normalidad, elevación de las transaminasas y ausencia de lesiones histológicas que puedan sugerir otras etíologias. El sistema de puntuación debe ser aplicado cuando no se cumplan todas las condiciones anteriores y el diagnóstico no esté claro.
DIAGNÓSTICO DIFERENCIAL DE LA HAI
La hepatitis autoinmune debe diferenciarse de otras causas de hepatitis crónica. Los marcadores virales permiten la identificación de las hepatitis víricas, sin embargo , en ocasiones el diagnostico diferencial entre la hepatitis C y las HAI puede ser conflictivo. A fin de corroborar la presencia de una infección viral es necesario realizar pruebas confirmatorias como la detección de ARN.
El VHC muestra numerosas interacciones con el sistema autoinmune (46,47). La asociación de la hepatitis por VHC y la presencia de anticuerpos comunes a la HAI, fue inicialmente cuestionada ya que los sueros hipergammaglobulinémicos (como los de afectos de HAI) daban falsos positivos a las pruebas de primera generación para la detección de anticuerpos anti-VHC. Pero posteriormente, y con las pruebas de detección de ARN del VHC para el diagnóstico de la HCC ha sido bien documentada con una prevalencia tan variable como del 9-38% de ANA y 5-91% de SMA (aunque normalmente a títulos bajos) en enfermos por HCC, siendo infrecuente la presencia de anti-LKM-1 (0-10%) o de anti-LC1 (41,48). Sin embargo, esta asociación no se ha sido observada por otros autores (49), atribuyéndose esta discrepancia a la predisposición genética de la población estudiada.
Existe una cierta heterogeneidad geográfica en la prevalencia del VHC en los pacientes con anticuerpos anti-LKM positivos. Mientras que la infección por virus C en la hepatitis autoinmune tipo 2 es muy baja en Inglaterra (<2%), en Alemania y Francia está alrededor del 50%, y en Italia, España y Japón es del 90% (50-52).
Los anticuerpos LKM-1 asociados a HCC no reconocen las mismas secuencias antigénicas del CYP450 2D6 que los afectos de HAI y pueden reaccionar con antígenos microsomales todavía no bien identificados con un peso molecular entre 59 y 70 kDa (41).
La presencia de fenómenos autoinmunes asociados o inducidos por el virus C, está apoyado por la presencia de anticuerpos anti-GOR, que van dirigidos contra una proteína de fusión de un clon de DNA del VHC descrito por Mishiro (53). Se ha descrito anticuerpos específicos frente al pentadecapéptido GOR de síntesis hepática, anti-GOR, que se encuentra en casi el 80% de los sueros de pacientes con HC. Sin embargo, los anti-GOR parecen tratarse más bien de anticuerpos dirigidos frente a el core del VHC que muestran reacción cruzada con los autoantígenos nucleares, antes que autoanticuerpos propiamente dicho. La prevalencia de anti-GOR varía según la positividad de los anti-LKM-1 y anti-VHC; si ambos son positivos los anti-GOR se detectan en el 80% de los pacientes, pero la proporción baja al 50% si sólo es positivo el anti-VHC y se cifra en un escaso 6% cuando sólo se encuentra presente el anti-LKM-1 (42).
Se llegó a proponer una división del tipo 2 de HAI (2a y 2b), basándose en su asociación o no con el VHC. No obstante, actualmente la presencia de estos anticuerpos en pacientes afectos de HCC parece considerarse más bien, como un epifenómeno (excepto en aquellos muy raros casos de hepatitis por coexistencia de HAI con VHC que no debe afectar al tratamiento establecido para esta hepatitis vírica (41). Es importante el diagnóstico diferencial entre HAI y HCC y la clasificación del paciente en uno u otro grupo tiene importantes connotaciones bajo el punto de vista terapéutico, puesto que un tratamiento erróneo con IFN-α en un paciente afecto de HAI puede exacerbar la enfermedad y uno corticoideo en un enfermo con HC puede incrementar la replicación del virus (54). La respuesta a IFN-α en los pacientes afectos de HCC con autoanticuerpos comunes a la HAI es similar a aquellos que carecen de ellos (55).
En el diagnostico diferencial de las HAI es importante descartar la ingesta importante de alcohol y de algunos medicamentos, ya que estos pacientes pueden presentar características clínicas y histológicas de hepatitis crónica difíciles de distinguir de la HAI.
La CBP y la CEP constituyen afecciones autoinmunes hepáticas aunque las estructuras diana del proceso no son los hepatocitos sino los colangiocitos de los conductos biliares. La CBP se presenta en mujeres mayores con colestasis crónica, elevación de la fosfatasa alcalina, lesión de los conductos biliares de pequeño tamaño y anticuerpos antimitocondriales positivos.
En cambio, la CEP se presenta preferentemente en hombres, se asocia a rectocolitis ulcerosa y hay lesión de los conductos biliares de mayor tamaño tanto intra como extra hepáticos.
Cuando los mecanismos de auto-tolerancia inmunitaria fracasan y el hígado se convierte en un órgano diana para el sistema inmunitario, la autodestrucción puede dirigirse tanto a los hepatocitos (HAI) como a las células epiteliales de los conductos biliares: CBP y CEP. Existen pacientes (7-20% de las HAI) en los que parecen afectarse simultáneamente tanto los hepatocitos como células biliares, y en los que el diagnóstico diferencial con la CBP o CEP puede ser difícil ya que cumplen criteros de HAI y a su vez muestran alteraciones propias de posible afectación ductal (39). Así pues, se puede definir el síndrome de solapamiento también conocido como colangitis autoinmune o CBP con AMA negativos, como aquél que cumpliendo criterios de HAI presenta a su vez signos de una de las dos enfermedades colestásicas autoinmunes: CBP o CEP (56).
El diagnóstico de la CBP se basa en la demostración de los anticuerpos antimitocondriales (AMA) y en concreto, aquellos dirigidos contra la subunidad E2 del complejo piruvato deshidrogenasa de la membrana interna mitocondrial (M2). La sensibilidad y especificidad diagnósticas de los anti-M2 es superior al 95%. También es característico de la CPB la presencia de hipergammaglobulinemia IgM.
En aquellos pacientes con HAI sin síndrome colestásico (objetivable bioquímica e histológicamente) ni hipergamma-globulinemia IgM pero con anti-M2, deberemos realizar un seguimiento pues es probable que acaben desarrollando una CBP (39).
Un grupo de pacientes pueden presentar síndrome colestásico con características de CBP pero no presentan anticuerpos antimitocondriales, pueden presentar otros anticuerpos, como ANA y/o SMA, y responden bien al tratamiento con esteroides. Esta situación mas que como un síndrome de sobreposicion de CBP y HAI, ha sido definida como una "colangitis autoinmune" (57).
A diferencia que en el solapamiento HAI/CBP, la CEP afecta predominantemente a hombres y se asocia con una enfermedad inflamatoria vesicular en aproximadamente el 70% de los casos. El diagnóstico de la CEP se basa en hallazgos bioquímicos de colestasis, presencia serológica de anti-p-ANCA (que también se pueden encontrar en la HAI) y cambios colangiográficos típicos. Se ha sugerido recientemente que el síndrome de solapamiento HAI/CEP no es sino un proceso evolutivo a partir de una HAI colestásica hacia una CEP, considerándolo como un síndrome secuencial (58).
Raramente, puede producirse un solapamiento entre la HAI y la hepatitis asociada a LES, en este caso la determinación de DNA, que suele ser negativa en el primer caso y positiva en el LES activo, ayudaría a discernir la entidad patológica.
ESTUDIOS DE IMAGEN
En general, no son útiles para alcanzar un diagnóstico definitivo. Sin embargo, la presencia de una ecotextura en la ultrasonografía o un anormal incremento de contraste en un TAC abdominal pueden sugerir la presencia de inflamación activa o necrosis. La observación de un hígado nodular irregular puede confirmar la presencia de cirrosis que es un importante factor pronóstico. Además, estos estudios de imagen pueden ser usados para descartar la presencia de un carcinoma hepatocelular (59).
EPIDEMIOLOGÍA
Mucha de la información epidemiológica sobre la HAI se ha basado en estudios previos sobre la hepatitis crónica activa. Sólo existe un estudio epidemiológico sobre la HAI en el cual se ha excluído a los enfermos con hepatitis C, y pese a que no está basado en los criterios dignósticos del GIHA, es el más fiable de los realizados hasta ahora. Este estudio, realizado en Noruega, establece, la incidencia anual media entre los individuos de raza blanca del norte de Europa es del 1.9/100.000 habitantes y la prevalencia de 16.9/100.000 habitantes (60). Sin embargo, en Japón la prevalencia estimada es sólo de 0.08-0.015/100.000 habitantes. La relación entre mujeres y hombres es de 4-6:1 (no obstante es más frecuente en los varones cuando la población es de avanzada edad ) (60). En EE.UU. la frecuencia de HAI entre los pacientes con enfermedades hepáticas crónicas supone entre el 11-23%, pero en Brasil supone 5-10% de las mismas (56).
La enfermedad es susceptible de adquirirse a todas las edades. Aunque, según algunos autores existe una distribución bifásica con dos picos de incidencia: uno el peripuberal y el otro entre la tercera y sexta década de vida (61), correspondiendo el segmento de edad mayoritario entre 50-70 años, otros autores no encuentran estas diferencias (60).
La HAI tipo 1 (95% de las HAI) es predominante en adultos entre 30 y 60 años y es la mayoritaria, la tipo 2 (5% de las HAI) es más prevalente en niños y mujeres adolescentes .
Un reciente estudio muestra que los afroamericanos afectos de HAI tienen mayor probabilidad para el desarrollo de cirrosis y precisan de mayor dosis de inmunosupresores para alcanzar la remisión que los caucásicos, sin embargo el pronóstico a largo plazo en ambas razas es similar (60).
Se estima que existen entre 100.000 y 200.000 personas afectas de HAI en los Estados Unidos y que está presente en el 2.6% de los receptores de trasplantes hepáticos en Europa y el 5.9% en EE.UU.
La historia natural propia de las hepatitis crónicas, de progresión a cirrosis y hepatocarcinoma, también es aplicable a la HAI, aunque la frecuencia parece ser menor que la de las hepatitis crónicas virales. En algunos casos esta progresión se asocia a la coexistencia con infección por VHC por lo que diversos autores consideran que no existen evidencias que permitan afirmar que la HAI comporta un riesgo adicional para el desarrollo de hepatocarcinoma (62).
CLÍNICA
La mayoría de los afectados son mujeres (70%). La clínica puede variar, dentro de un amplio espectro, desde una hepatitis subclínica asintomática (pero con resultados analíticos anormales) hasta una hepatitis aguda que suele ser de tipo 2 y que en algunos casos se resuelve espontáneamente.
La presentación aguda se da hasta en un 30-40% de los pacientes, a menudo con encefalopatía, franca ictericia y marcada coagulopatía. Su rápido diagnóstico y terapia es fundamental para evitar la progresión a fallo hepático subagudo que pudiera requerir un trasplante hepático. Pero la presentación más frecuente es como enfermedad hepática crónica (43).
El inicio de la enfermedad suele ser insidioso (en un 50% de los pacientes) , con síntomas inespecíficos pero frecuentes de las hepatopatías crónicas, un tercio de los pacientes refieren molestias en el hipocondrio derecho. Una vez instaurada la enfermedad, los síntomas más comunes, compartidos con la mayoría de afecciones hepáticas son: hepatomegalia (83%), ictericia (69%), arañas vasculares (58%) y esplenomegalia (32%). Con frecuencia los únicos síntomas que presente el paciente son dermatológicos o reumáticos, éstos suelen presentar una enfermedad más benigna que responde mejor y con más prontitud a la terapéutica inmunosupresora que aquellos en los que se observa una presentación aguda o insidiosa (63). Los que muestran una cirrosis claramente establecida manifiestan las complicaciones clásicas de ésta (40,59).
Aproximadamente, un 20% presentan inicialmente signos de descompensación cirrótica. Otros evolucionan a una hepatitis rápidamente progresiva después de años de tratamiento infructuoso marcado por múltiples recidivas.
La HAI se asocia con una gran variedad de desórdenes de origen inmunológico en hasta un 40% de los pacientes (60,63).
La existencia de HAI de inicio agudo como entidad diferenciada ha sido controvertida, de hecho algunos autores no encuentran sustanciales diferencias y la consideran meramente como una exacerbación de la forma crónica. Sin embargo, otros aprecian diferencias entre ambas presentaciones, así objetivan concentraciones de ALT significativamente superiores al resto de la HAI, menor nivel de IgG, afección histopatológica con mayor prevalencia de necrosis centrolobulillar e infiltración predominante de linfocitos CD8 en la hepatitis de interfase, y menor estadiaje y puntuación (según criterios GHIA) (64).
Hay que tener presente que la titulación de los anticuerpos no es un indicador completamente fiable de la actividad de la enfermedad (65).
HEPATITIS EN EL SÍNDROME DE POLIGLANDULAR AUTOINMUNE TIPO 1
La hepatitis se observa en un 10-15% de los pacientes con Síndrome Poliglandular Autoinmune tipo 1 (SPA-1). Este raro síndrome se caracteriza por candidiasis mucocutánea, hipotiroidismo, enfermedad de Addison y otras endocrinopatías. En estos pacientes, los autoanticuerpos frente a CYP450 1A2 se han descrito como marcadores de enfermedad hepática (Tabla II) (56).
PRESENTACIÓN PEDIÁTRICA
La presentación pediátrica tiene un amplio espectro clínico que va desde la elevación de aminotransferasas asintomática hasta la forma grave progresiva. Al igual que en los adultos, se reconocen dos tipos de HAI, según la presencia de anticuerpos anti- ANA/SMA (tipo I) o anti-LKM1 (tipo 2). Las series pediátricas muestran similar severitud en ambos tipos de hepatitis y una prevalencia mucho mayor en niñas (75%).
El tipo 2 suele presentarse en forma aguda, a edad más temprana y con frecuente deficiencia en IgA, mientras que no se observan diferencias en la clínica, historia familiar, presencia de desórdenes autoinmunes asociados o respuesta al tratamiento (66). Ocasionalmente, puede desarrollar un fallo hepático agudo.
En un 50% de los tipo 1 y en un 65% de los tipo 2, la presentación es indistinguible de la hepatitis viral aguda con síntomas inespecíficos de hepatitis.
Las características serológicas de la HAI en niños difiere sustancialmente a la de los adultos. Un 38% de los niños presentan anticuerpos anti-LKM1 (tipo 2), al contrario que los adultos en los que sólo se observan en un 3-4%. En la tipo 2 (62%), el título de SMA puede ser bajo (1:40). Del 50-65% muestran una presentación aguda, a menudo fulminante; y alrededor del 69% de los niños con tipo 1 y el 38% del tipo 2 ya presentan fenómenos cirróticos en su inicio. La edad media de debut en niños es a los 10 años.
Al igual que en los adultos, es frecuente la coexistencia con otras enfermedades autoinmunes (67). El tipo más común de CEP pediátrica es la autoinmune. La clínica, bioquímica, inmunología e histología es indistinguible de la HAI, el diagnóstico se realiza por colangiografía. Existen dudas sobre si se trata de una entidad diferenciada de la HAI o de un tipo de presentación de esta.
PRESENTACIÓN OBSTÉTRICA
La presentación de la HAI en mujeres embarazadas en infrecuente ya que como hemos dicho anteriormente, una de la sintomatología propia de la enfermedad es la amenorrea. En la escasa literatura sobre el tema se coincide en afirmar que la gestación no empeora , en general, el diagnóstico de la enfermedad, aunque en un 20% de las gestantes aparecen complicaciones, la más importante la hemorragia de varices esofágicas y el sangrado vaginal. Aunque sí que se observa una elevada tasa de pérdida fetal (30-64,5%) y un aumento de la prematuridad (18-30%). Por tanto, un diagnóstico temprano de la enfermedad es crucial para evitar riesgos fetales (40).
TRATAMIENTO
Sin tratamiento el 40% de los pacientes afectos de HAI severa fallecían durante los 6 meses posteriores al diagnóstico y un 40% de los supervivientes desarrollan cirrosis, la tasa de mortalidad a los cinco años alcanzaba el 80% (40). El objetivo del tratamiento es la completa resolución bioquímica e histológica de la inflamación hepática, así como la remisión de los síntomas clínicos. Todos los pacientes deben ser tratados independientemente del tipo de HAI, forma de presentación o severidad de la enfermedad. Los síntomas, potenciales efectos secundarios y comportamiento de la enfermedad deben ser sopesados para un tratamiento individualizado cuando la afección es moderada o leve (68). La medicación debe ser mantenida hasta que se muestre remisión, fallo en el terapia, intolerancia farmacológica o una respuesta incompleta. Dado el escaso número de curaciones (10%) y el elevado de recidivas, el objetivo terapéutico debe fijarse en la dosis mínima eficaz que mantenga la remisión, siendo el mejor índice de respuesta al tratamiento los niveles de aminotransferasas que deben mantenerse por debajo de 2 veces el límite superior de referencia (69). Los fármacos inmunosupresores disminuyen la mortalidad en la HAI, aunque no se sabe con certeza si previene o enlentece la progresión a cirrosis (38).
El tratamiento se basa en corticoesteroides sólo o en combinación con azatioprina (de la que no existe evidencia de ongenicidad con las dosis habituales). La tasa de remisión global puede alcanzar el 80%. Con tratamiento, las expectativas de supervivencia son: 94%, 90% y superiores al 80%, a 5, 10 y 20 años, respectivamente. Aunque se desconoce si previenen o enlentecen la progresión a cirrosis (69).
El examen histológico garantiza la remisión antes de la retirada farmacológica cuando hay ausencia de sintomatología y las pruebas de laboratorio son normales o cercanas a la normalidad. Cuando no existe respuesta al tratamiento se puede intentar con terapia a dosis alta. La toxicidad farmacológica o una respuesta incompleta pueden soslayarse mediante un ajuste individualizado de terapia. Bajas dosis de corticosteroides (2.5-10 mg/día) o terapia indefinida con azatioprina (2 mg/kg/día) están indicadas en pacientes que presentan recidivas múltiples (un 47% de los pacientes presentan una recidiva en un seguimiento a diez años) (70).
Previamente a la administración de azatioprina se debe determinar la actividad de la tiopurina metiltransferasa (TPMT) y ajustar la dosis en aquellos pacientes con baja actividad enzimática, obviando el riesgo de citopenia ocasionado por la 6-mercaptopurina (71).
El tratamiento con corticosteroides se asocia a los efectos secundarios clásicos, incluso a pueden producir atrofia muscular y predisposición a infecciones.
Según Czaja el tratamiento está indicado en las condiciones que se enumeran en la tabla III (72). Desde un punto de vista práctico pueden diferenciarse dos tratamientos: uno dirigido a alcanzar la remisión de la enfermedad y otro para el mantenimiento. Que a su vez, se pueden subclasificar en monoterapia o terapia de combinación (Fig. 4) (73). En pacientes jovenes con actividad inflamatoria baja (amino-transferasas ≤ 5 veces el límite superior de referencia) se puede obviar la terapia de remisión comenzando con dosis de mantenimiento (56).


TERAPIA DE REMISIÓN
La dosificación dependerá según la presentación (aguda/crónica) y el daño histológico. Tiene por objeto conseguir la remisión (o respuesta completa) definida según criterios establecidos por Grupo Internacional para la Hepatitis Autoinmune (Tabla IV) .

Monoterapia. Existe consenso para considerar como tratamiento de elección a los glucocorticoides sintéticos de primera generación: prednisona, prednisolona o metilprednisolona. El daño hepático en la cirrosis avanzada no suele ser suficiente para afectar a la metabolización de la prednisona a prednisolona (metabolito activo) y no justifica la administración preferente de esta última. La azatioprina no es eficaz como monoterapia para conseguir la remisión (56).
Presentación aguda y actividad histológica severa. Se aconseja corticosteroides en dosis de choque de 1 mg/kg/día (hasta un máximo de 60 mg/día) durante 1-2 semanas, reduciendo gradualmente hasta 10-15 mg/día una vez alcanzada la remisión
HAI asintomática con leve actividad histológica. Dosis de 20-30 mg/día durante 1-2 semanas con gradual reducción hasta 5 mg/día en remisión.
Combinada. Esta es la terapia recomendada. La asociación con azatioprina permite la reducción de la dosis de corticosteroides disminuyendo sus efectos colaterales (10% frente al 44% en la monoterapia).
Presentación aguda y actividad histológica severa. Corticosteroides a dosis de 30-60 mg/día + azatioprina a dosis de 50-100 mg/día durante 1- 2 semanas. Reducción gradual de la dosis de los corticosteroides hasta 10 mg/día manteniendo la dosis de azatioprina hasta alcanzar la remisión.
HAI asintomática con leve actividad histológica. Corticosteroides a dosis de 15 mg/día + azatioprina a dosis de 50 mg/día durante 1- 2 semanas. Posteriormente se reducen gradualmente la dosis de los corticosteroides hasta 5 mg/día manteniendo la dosis de azatioprina hasta alcanzar la remisión.
TERAPIA DE MANTENIMIENTO
Aunque la remisión puede ser duradera, las recidivas son frecuentes al retirar el tratamiento. Más del 70% de los pacientes con HAI requieren terapia inmunosupresora de por vida. La terapia de mantenimiento tiene por objetivo evitar la recidiva, definida según criterios establecidos (Tabla IV) de la enfermedad, utilizando la mínima dosis de inmunosupresores y reducir al mínimo los efectos colaterales y secundarios del tratamiento. Las concentraciones de aminotransferasas el mejor parámetro para monitorizar el mantenimiento de la remisión. En aquellos casos de HAI leve (10-30% de los pacientes) que muestran normalización de los parámetros bioquímicos e histológicos y después de un mínimo de 2-4 años de tratamiento, la terapia puede interrumpirse siempre de forma gradual ya que una interrupción drástica puede ocasionar una exacerbación de la enfermedad.
Monoterapia. Se aconseja una dosis de mantenimiento con corticosteroides de 2.5-10 mg/día o bien con azatioprina de 50-100 mg/día. Este último tratamiento es el preferido por la menor frecuencia de efectos secundarios y colaterales, incluso hasta dosis de 2 mg/kg/día.
Combinada. Si el tratamiento monoterápico con azatioprina no es capaz de mantener una remisión estable se recomienda asociarlo a corticosteroides 5-7,5 mg/día.
TRATAMIENTO PEDIÁTRICO
El tratamiento debe ser garantizado a la mayor parte de los niños en el momento de su diagnóstico (68). La respuesta a los inmunosupresores alcanza hasta el 80%, con un 60-80% de remisiones. Los corticosteroides son la terapia de elección, aunque no se ha alcanzado consenso sobre la dosis inicial, la forma farmacéutica o si debe ser monoterapia o asociada a azatioprina.
La monoterapia se basa en un tratamiento de choque durante dos semanas con la administración de 1-2 mg/kg/día de corticosteroides con un máximo de 60 mg/día y una reducción gradual a dosis de mantenimiento de 5-10 mg/día.
Se prefiere el tratamiento de mantenimiento diario con corticosteroides con dosis mínima, dado la mala cumplimentación de los pacientes, especialmente de los adolescentes (67). Debido a los efectos secundarios del tratamiento con corticosteroides sobre el crecimiento óseo es aconsejable una pronta asociación con azatioprina a dosis de 1-2 mg/kg/día, o incluso 6-mercaptopurina.
La duración óptima del tratamiento no está definida, se ha descrito que en una cuarta parte de los pacientes pediátricos con tipo HAI 1 se pudo retirar la medicación (previa biopsia hepática) después de un año de función hepática normal, pero no ocurrió lo mismo con los de grupo HAI 2. No existen estudios controlados en niños, por lo que la decisión de retirada del medicamento debe ser valorada cuidadosamente de forma individual (67).
TRATAMIENTO OBSTÉTRICO
Aunque no se ha demostrado que el tratamiento inmunosupresor sea teratogénico, se aconseja rebajar las dosis de corticosteroides y azatioprina a las mínimas eficaces. Es preciso evitar la lactancia materna porque ambos fármacos se excretan por leche.
OTROS TRATAMIENTOS
Aproximadamente un 10% de los pacientes afectos de HAI no muestran respuesta al tratamiento inmunosupresor con corticoides de primera generación, otro 15% sólo responden parcialmente y en un 13% el tratamiento debe interrumpirse a causa de los efectos secundarios; por tanto, se debe recurrir a tratamientos alternativos en un significativo número de casos.
Tratamientos inmunosupresores que se han utilizado para los casos refractarios o la intolerntesa los corticosteroides son: ciclosporina, budesonida, tacrolimus, micofenolato mofetilo, ácido ursodeoxicólico (cuando se asocia a colestasis), metotrexato, ciclofosfamida, mercaptopurina, deflazacort, gammaglobulina intravenosa, fosfatidilcolina, tiazolidina carboxilato, broqueinar, ripamicina y depuradores de radicales libres.
También existen nuevas terapias inmunosupresoras, actualmente en fase de experimentación, que pudieran ser eficaces en un futuro próximo y que podrían incluir a: anticuerpos bloqueadores frente al TNF, péptidos inhibidores competitivos dirigidos al complejo receptor de células T- CMH clase II, antígeno 4 linfocitario T citotóxico soluble, citoquinas (IL-10) vacunación con células T, vacunación o anticuerpos contra el producto protéico del gen de célula T (TIRC7), y la terapia génica (74).
En casos de cirrosis avanzada, ascitis intratable, encefalopatía hepática intrínseca, varices esofágicas hemorrágicas o refractariedad o intolerancia a los tratamientos anteriores, se puede plantear la necesidad de un trasplante hepático ortotópico. En Europa un 4% de los pacientes cirróticos con diagnóstico de HAI son tributarios de trasplante (75). La tasa de supervivencia del trasplante hepático es excelente (83-92% a los cinco años, 75% a los 10 años) con negativización serológica a los autoanticuerpos dentro de los dos años posteriores. Aunque existe un riesgo de recurrencia de hasta un 10-60% por reactivación de las células T de memoria por antígenos específicos a través de un mecanismo que no requiera compatibilidad CMH; esta reactivación raramente progresa a cirrosis o fallo del injerto. La HAI raramente recidiva durante el primer año, posiblemente a causa de las altas dosis de inmunosupresores a que se someten los pacientes. Algunos autores consideran los criterios de puntuación del GIHA como demasiado estrictos para diagnosticar una recurrencia en trasplantados, opinan que el diagnóstico se debe sustentar en alteraciones bioquímicas mantenidas y signos clínicos. La recurrencia es mucho menor en HAI tipo 2 (5%) que en la de tipo 1( 34%) (76). Diferentes autores han comprobado que una mejor pronóstico postrasplante en aquellos pacientes diagnosticados incialmente de HAI aguda fulminante/subfulminante, y dentro de este grupo los que muestran necrosis hepática masiva frente a los que presentan necrosis, al compararlos con los de HAI crónica (77).
PROGNOSIS
Aquellos pacientes en los que se observa una completa normalización en la hepatitis tienen un riesgo muy bajo de recidivas (15-20%), sin embargo, si se presenta hepatitis de interfase el riesgo de recidiva es de un 90%. La curación completa se observa en sólo el 10% de los pacientes tratados, esta curación debe ser objetivada histopatológicamente. Un 21% muestra remisión de más de seis meses, presentan manifestaciones residuales aunque son funcionales y no son tributarios de nueva medicación. La normalización persistente de las aminotransferasas no implica necesariamente que no haya progresión a fibrosis, diferentes trabajos prospectivos han demostrado que de un 5-10% de estos pacientes acaban desarrollando cirrosis (37).
Son factores de mal pronóstico los valores de aspartato aminotranferasa de 5-10 veces el límite superior de referencia conjuntamente con hipergammaglobulinemia al menos dos veces el límite superior de referencia (mortalidad del 90% a los 10 años sin tratamiento) (68). De los pacientes con hepatitis periportal, la cirrosis se desarrolla en el 17% a los 5 años, pero alcanza al 82% cuando existen puentes de necrosis o necrosis multiacinar (56). La presencia de cirrosis se asocia a una mortalidad del 58% a los 5 años, sin embargo no influye en la respuesta o el resultado a corto plazo del tratamiento si está compensada. Sólo el 1% de los cirróticos, en un seguimiento a diez años, generan hepatocarcinoma (68).
Se ha descrito que los pacientes con alelo nulo en el locus C4 presentan una mayor frecuencia de recidiva e incremento de mortalidad (40).
El desarrollo de la HAI está también influenciado significativamente por el perfil HLA. La presencia del haplotipo HLA B8 se asocia con inflamación en el debut y con mayor frecuencia de recidiva una vez finalizado el tratamiento. Los pacientes con haplotipo HLA DR3 se caracterizan por la dificultad para alcanzar la remisión, presentar mayor tasa de recurrencia y de trasplante. En los HLA DR4, por su parte, se suele observar un debut a mayor edad y un desarrollo más benigno de la enfermedad.
AGRADECIMIENTOS
Al Dr. E. Orfei (Loyola University, Chicago), al Dr. M. Tagle (Universidad Cayetano Heredia, Lima) por sus consentimientos en la reproducción de las fotografías, y al Dr. R. Cano (Hospital de la Ribera, Alzira) por sus comentarios histopatológicos.
Bibliografía
1. Johnson PJ, McFarlane IG. Meeting report: International Autoimmune Hepatitis Group. Hepatology 1993; 18:998-1005. [ Links ]
2. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis J Hepatol 1999; 31:929-938. [ Links ]
3. Robertson DAF, Zhang SL, Guy EC, Wright R. Persistent measles virus genome in autoimmune chronic active hepatitis. Lancet 1987; 2: 9-11. [ Links ]
4. Vento S, Garofano T, Di Perri G, Dolci L, Concia E, Bassetti D. Identification of hepatitis A virus as a trigger for autoimmune chronic hepatitis type 1 in susceptible individuals. Lancet 1991; 337: 1183-1187. [ Links ]
5. Tagle Arrospide M, León Barúa R. Hepatitis viral A como factor desencadenante de hepatitis autoinmune. Reporte de un caso y revisión de la literatura. Rev Gastroenterol Peru 2003; 23 : 134-137. [ Links ]
6. Laskus T, Slusarczyck J. Autoimmune chronic active hepatitis developing after acute type B hepatitis. Dig Dis Sci 1989; 34: 1294-1297. [ Links ]
7. Berk L, Schalm SW, Heijtink RA. Severe chronic active hepatitis (autoimmune type) mimicked by coinfection of hepatitis C and human immunodeficiency viruses. Gut 1991; 32: 1198-1200. [ Links ]
8. Silva E, Sallie R, Tibbs C, McFarlane I, Johnson P, Williams R. Absence of hepatitis C virus in British patients with type I autoimmune chronic active hepatitis - a polymerase chain reaction and serological study. J Hepatol 1993; 19: 211-215. [ Links ]
9. Medina J, Caveda L, Sanz-Cameno P, et al. Hepatocyte growth factor activates endothelial proangiogenic mechanisms relevant in chronic hepatitis C-associated neoangiogenesis. J Hepatol 2003; 38: 660-667. [ Links ]
10. Manns MP, Obermayer-Straub P. Triggering factors and immunogenetics of autoimmune hepatitis. In: Moreno-Otero R, Clemente-Ricote G, García-Monzón C, eds. Immunology and the liver: autoimmunity. Madrid: Arán Ediciones S.A., 2000. [ Links ]
11. Prandota J. Possible pathomechanism of autoimmune hepatitis. Am J Ther 2003; 10: 51-57. [ Links ]
12. García-Buey L, García-Monzón C, Rodríguez S. et al. Latent autoimmune hepatitis triggered during interferon therapy in patients chronic hepatitis C. Gastroenterology 1995; 108: 1770-1777. [ Links ]
13. Schatnner A. Interferon and autoimmunity. Am J Med Sci 1988; 295: 532-544. [ Links ]
14. Van den Oord JJ, De Vos R, Desmet VJ. HLA expresión in liver disease. Prog Liver Dis 1990; 9: 73-88. [ Links ]
15. Manns MP, Obermayer-Straub P. Cytochromes P450 and uridine triphosphate-glucuronosyltransferases: model autoantigens to study drug-induced, virus-induced, and autoimmune liver disease. Hepatology 1997; 26: 1054-1066. [ Links ]
16. Stockert RJ. The asialoglycoprotein receptor: relationships between structure, function and expression. Physiol Rev 1995; 75: 591-609. [ Links ]
17. Wies I, Brunner S, Henninger J, et al. Identification of target antigen for SLA/LP autoantibodies in autoimmune hepatitis. Lancet 2000; 355: 1475-1476. [ Links ]
18. Costa M, Rodríguez-Sánchez JL, Czaja AJ, Gelpi C. Isolation and characterization of cDNA encoding the antigenic protein of the human tRNP(Ser)Sec complex recognized by autoantibodies from patients with type-1 autoimmune hepatitis. Clin Exp Immunol 2000; 121: 364-374. [ Links ]
19. Pelli N, Fensom AH, Slade C, Boa F, Mieli-Vergani G, Vergani D. Argininosuccinate lyase: a new autoantigen in liver disease. Clin Exp Immunol 1998; 114: 455-461. [ Links ]
20. Lapierre P, Hajoui O, Homberg JC, Alvarez F. Formiminotransferase cyclodeaminase is an organ-specific autoantigen recognised by sera of patients with autoimmune hepatitis. Gastroenterology 1999; 116: 643-649. [ Links ]
21. Volpes R, van den Oord JJ, Desmet VJ. Immunohistochemical study of adhesión molecules in liver inflammation. Hepatology 1990; 12: 59-65. [ Links ]
22. Austchbach F, Meuer SC, Moebius U, et al. Hepatocellular expression of lymphocyte function-associated antigen 3 in chronic hepatitis. Hepatology 1991; 14: 223-230. [ Links ]
23. Tigl H, Wilmer A, Vogel W, et al. Serum levels of cytokines in chronic liver diseases. Gastroenterology 1992; 103: 264-274. [ Links ]
24. Al-Janadi M, Al-Wabel A, Raziuddin S. Soluble CD3 and interleukin-4 levels in autoimmune chronic active hepatitis and systemic lupus erythematosus. Clin Immunol Immunopathol 1994; 71: 33-37. [ Links ]
25. Schlaak JF, Lohr H, Gallati H, Meyer zum Buschenfelde KH, Flescher B. Analysis of the in vitro cytokine production by liver-infiltrating T cells of patients with autoimmune hepatitis. Clin Exp Immunol 1993; 94: 168-173. [ Links ]
26. Nishioji k, Okanoue T, Itoh Y, et al. Increase of chemokine interferon-inducible protein-10 (IP-10) in the serum of patients with autoimmune liver diseases and increase of its mRNA expression in hepatocytes. Clin Exp Immunol 2001; 123: 271-279. [ Links ]
27. Sanz-Cameno P, Medina J, Garcia-Buey L, et al. Enhanced intrahepatic inducible nitric oxide synthase expression and nitrotyrosine accumulation in primary biliary cirrhosis and autoimmune hepatitis. J Hepatol 2002; 37: 723-729. [ Links ]
28. Czaja AJ, Santrach PJ, Moore SB. Shared genetic risk factors in autoimmune liver disease. Dig Dis Sci 2001; 46: 140-147. [ Links ]
29. Czaja AJ, Doherty DG, Donaldson PT. Genetic Bases of Autoimmune Hepatitis. Dig Dis Sci 2002; 47: 2139-2150. [ Links ]
30. Whitacre CC, Reingold SC, O'Looney PA. A gender gap in autoimmunity. Science 1999; 283: 1277-1278. [ Links ]
31. Stretell MDJ, Donaldson PT, Thompson LJ, Santrach PJ, et al. Allelic basis for HLA-encoded susceptibility to type 1 autoimmune hepatitis. Gastroenterology 1997; 112: 2028-2035. [ Links ]
32. Czaja AJ, Kruger M, Santrach PJ, et al. Genetic distintions between types 1 and 2 autoimmune hepatitis. Am J Gastroenterol 1997; 92: 2197-2200. [ Links ]
33. Czaja AJ. Understanding the pathogenesis of autoimmune hepatitis. Am J Gastroenterol 2001; 96: 1224-1231. [ Links ]
34. Donaldson PT. Genetics in autoimmnue hepatitis. Semin Liver Dis 2002; 22: 353-364. [ Links ]
35. Cookson S, Constatini PK, Clare M, et al. Frequency and nature of cytokine gene polymorphisms in type 1 autoimmune hepatitis. Hepatology 1999; 30: 851-856. [ Links ]
36. Agarwal K, Jones DE, Daly AK, et al. CTLA-4 gene polymorphism confers susceptibility to primary biliary cirrhosis. J Hepatol 2000; 32: 538-541. [ Links ]
37. Luxon BA. Autoimmune hepatitis. Making sense of all those antibodies. Postgrad Med 2003; 114: 79-82; 85-88. [ Links ]
38. Moreno Otero R. Hepatitis autoinmune. Medicine 2000; 08: 693-701. [ Links ]
39. Dienes HP, Erberich H, Dries V, Schirmacher P, Lohse A. Autoimmune hepatitis and overlap syndromes. Clin Liver Dis 2002; 6: 349-362. [ Links ]
40. McFarlane IG. Definition and classification of autoimmune hepatitis. Semin Liver Dis 2002; 22: 317-324. [ Links ]
41. Dalekos GN, Zachou K, Liaskos C, Gatselis N. Autoantibodies and defined target autoantigens in autoimmune hepatitis: an overview. Eur J Intern Med 2002; 13:293-303. [ Links ]
42. Strassburg CP, Manns MP. Autoantibodies and autoantigens in autoimmune hepatitis. Semin Liver Dis 2002; 22: 339-352. [ Links ]
43. Krawitt EL. Autoimmune hepatitis. N Engl J Med. 1996; 334: 897-903. [ Links ]
44. Zauli D, Ghetti S, Grassi A, Descovich C, Cassani F, Ballardini G et al. Anti-neutrophil cytoplasmic antibodies in type 1 and 2 autoimmune hepatitis. Hepatology 1997; 25: 1105-1107. [ Links ]
45. Roozendaal C, de Jong MA, van den Berg AP, van Wijk RT, Limburg PC, Kallenberg CG. Clinical significance of anti-neutrophil cytoplasmic antibodies (ANCA) in autoimmune liver diseases. J Hepatol 2000; 32: 734-741. [ Links ]
46. Ramos-Casals M, Font J, Ingelmo M. Prevalencia y significado clínico de la infección crónica por el virus de la hepatitis C en las enfermedades autoinmunes sistémicas. Med Clin (Barc) 2001; 116: 701-709. [ Links ]
47. Strassburg CP, Vogel A, Manns MP. Autoimmunity and hepatitis C. Autoimmun Rev 2003; 2: 322-331. [ Links ]
48. Zauli D, Cassani F, Bianchi FB. Auto-antibodies in hepatitis C. Biomed Pharmacother 1999; 53: 234-241. [ Links ]
49. Sachithanandan S, Fielding JF. Autoimmune disease is not a feature of hepatitis C infection in Ireland. J Clin Gastroenterol 1997; 25: 522-524. [ Links ]
50. Lenzi M, Johnson PJ, Mcfarlane IG et al. Antibodies to hepatitis C virus in autoimmune liver disease: evidence for geographical heterogeneity. Lancet 1991; 338: 277-280. [ Links ]
51. Michel G, Ritter A, Gerken G et al. Anti-GOR and hepatitis C virus in autoimmune liver diseases. Lancet 1992; 339: 267-269. [ Links ]
52. Lunel F, Abuaf N, Frangeul L et al. Liver-kidney microsome antibody type I and hepatitis C virus infection. Hepatology 1992; 16: 630-636. [ Links ]
53. Mishiro S, Hoshi Y, Takeda K et al. Non-A non-B specific antibodies directed at host-derived epitope: implication for an autoimmune process. Lancet 1990; 336: 1400-1403. [ Links ]
54. Jiménez J, Llorente MJ. Anticuerpos contra las mitocondrias y otros orgánulos subcelulares. En: González de Buitrago, González Rodríguez, directores. Enfermedades autoinmunitarias y autoanticuerpos. Barcelona: Monografías del Comité de Publicaciones de la Sociedad Española de Bioquímica Clínica y Patología Molecular, 2000. p. 129-156. [ Links ]
55. Cassani F, Cataleta M, Valentini P, Muratori P, Giostra F, Francesconi R, Muratori L, Lenzi M, Bianchi G, Zauli D, Bianchi FB. Serum autoantibodies in chronic hepatitis C: comparison with autoimmune hepatitis and impact on the disease profile. Hepatology. 1997; 26:561-516. [ Links ]
56. Manns MP, Strassburg CP. Autoimmune hepatitis: Clinical Challenges. Gastroenterology 2001; 120: 1502-1517. [ Links ]
57. Lohse AW, zum Buschenfelde KH, Franz B, Kanzler S, Gerken G, Dienes HP. Characterization of the overlap syndrome of primary biliary cirrhosis (PBC) and autoimmune hepatitis: evidence for it being a hepatitic form of PBC in genetically susceptible individuals. Hepatology 1999; 29: 1078-1084. [ Links ]
58. Abdo AA, Bain VG, Kichian K, Lee SS. Evolution of autoimmune hepatitis to primary sclerosing cholangitis: A sequential syndrome. Hepatology 2002; 36: 1393-1399. [ Links ]
59. Raghuraman UV. Autoimmune hepatitis. Disponible en: http://www.emedicine.com/MED/topic366.htm. [ Links ]
60. Feld J, Heathcote EJ. Epidemiology of autoimmune liver disease. J Gastroenterol Hepatol 2003; 18: 1118-1128. [ Links ]
61. Al-Khalidi JA, Czaja AJ. Current concepts in the diagnosis, pathogenesis, and treatment of autoimmune hepatitis. Mayo Clin Proc 2001; 76:1237-52. [ Links ]
62. Watanabe M, Moritani M, Hamamoto S, Uchida Y, Ishihara S, Adachi K, et al. Hepatocellular carcinoma complicating HCV-negative autoimmune hepatitis without corticosteroid therapy. J Clin Gastroenterol 2000; 30: 445-446. [ Links ]
63. Gómez de la Torre R, Florido Mancheño JI, Suárez del Villar R, Rubio Barbón S. Manifestaciones dermatológicas como forma de presentación de una hepatitis autoinmune. An Med Interna (Madrid) 2001; 18: 558-559. [ Links ]
64. Okano N, Yamamoto K, Sakaguchi K, Miyake Y, Shimada N, Hakoda T, et al. Clinicopathological features of acute-onset autoimmune hepatitis. Hepatol Res 2003; 25: 263-270 . [ Links ]
65. McFarlane IG. Autoimmune hepatitis: diagnostic criteria, subclassifications, and clinical features. Clin Liver Dis 2002; 6: 317-333. [ Links ]
66. Mieli-Vergani G, Vergani D. Autoimmune Liver Disease in children. Ann Acad Med Singapore 2003; 32: 239-43. [ Links ]
67. García Novo MD, Acuña D, Martínez-Gómez J, Pedrón C, Martínez Pérez A, de Vicente Prada I. Hepatitis autoinmune. Revisión de un serie de 11 casos pediátricos. An Esp Pediatr 2001; 54: 21-25. [ Links ]
68. Czaja AJ, Freese DK. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 2: 479-497. [ Links ]
69. Moreno R, García-Monzón C, García Buey L. Disponible en: http://www.aeeh.org/trat_enf_hepaticas/C-06.pdf. [ Links ]
70. Czaja AJ. Treatment of autoimmune hepatitis. Semin Liver Dis 2002; 22: 365-378. [ Links ]
71. Gisbert JP, González-Guijarro L, Cara C, Pajares JM, Moreno-Otero R. Actividad de la tiopurina metiltransferasa en pacientes con hepatitis autoinmune. Med Clin (Barc) 2003; 121: 481-484. [ Links ]
72. Czaja AJ. Treatment strategies in autoimmune hepatitis. Clin Liver Dis 2002; 6: 511-536. [ Links ]
73. Medina J, García-Buey L, Moreno-Otero R. Review article: Immunopathogenetic and therapeutic aspects of autoimmune hepatitis. Aliment Pharmacol Ther 2003; 17: 1-16. [ Links ]
74. Czaja AJ. Emerging treatments for autoimmune hepatitis. Curr Drug Targets Inflamm Allergy 2002; 1: 317-326. [ Links ]
75. Strassburg CP, Manns MP. Autoimmune hepatitis. Best Pract Res Clin Gastroenterol 2003; 17: 291-306. [ Links ]
76. Manns P, Matthias JB. Recurrent autoimmune hepatitis after liver transplantation-When non-self becomes self. Hepatology 2000; 32: 868-870. [ Links ]
77. Núñez-Martínez O, De la Cruz G, Salcedo M, Molina J, De Diego A, Ripoll C, et al. Liver transplantation for autoimmune hepatitis: fulminant versus chronic hepatitis presentation. Transplant Proc 2003; 35:1857-1858. [ Links ]
78. Coverdale SA, Field J, Farrell GC. How reversible is hepatic functional impairment in autoimmune hepatitis? J Gastroenterol Hepatol 2003; 18: 371-375. [ Links ]

