










 Custom services
Custom services
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La osteoporosis es una enfermedad compleja caracterizada por baja masa ósea y deterioro de la microarquitectura del tejido óseo que conduce a un mayor riesgo de fractura. Por ejemplo, en EE.UU. se producen 1,5 millones de nuevos casos de fracturas cada año, lo que representa una enorme carga económica para los sistemas de atención de la salud. La osteoporosis se define clínicamente a través de la medición de la densidad mineral ósea (DMO), que sigue siendo el mejor predictor de fractura [1,2]. Estudios de heredabilidad utilizando gemelos o familias han demostrado que el 50-85% de la variación en la densidad mineral ósea está determinada genéticamente [3]. Las fracturas osteoporóticas también muestran heredabilidad independiente de la densidad mineral ósea [4].
Los estudios de asociación en genoma completo (GWAs) han ampliado muchísimo la comprensión de la arquitectura genética de enfermedades comunes y complejas [5]. Esta aproximación genómica está proporcionando información clave sobre los mecanismos de la enfermedad, con perspectivas para el diseño de estrategias más eficaces de evaluación del riesgo de enfermedad y para el desarrollo de nuevas intervenciones terapéuticas [6]. Sin embargo, las variantes genéticas que se identifican en los GWAs se encuentran con frecuencia en las zonas no-codificantes del genoma, cuya posible función es menos conocida y en muchos casos estas señales pueden estar en desequilibrio de ligamiento con variantes causales que no han sido genotipadas. El meta-análisis de GWAs para DMO y fractura osteoporótica de Estrada et al. [7] identificó hasta 56 loci genómicos asociados con la DMO, 14 de los cuales también estaban asociados con las fracturas osteoporóticas. Uno de los SNPs cuya asociación con ambos fenotipos mostró una significación más sólida (rs4727338) se encuentra en una región intrónica del gen FLJ42280, señalándolo como un locus de susceptibilidad para la osteoporosis (Figura 1). Otros trabajos de GWA demostraron que otros SNPs intrónicos del mismo gen (rs7781370, rs10429035 y rs4729260) también se asociaban con la DMO [8,9]. FLJ42280 es un gen muy poco estudiado, cuya relación con la biología del hueso se desconoce.

Figura 1 Región genómica 7q21.3 con las señales de asociación a DMO de los SNPs testados por Estrada et al.[7].La coordenada entre la posición genómica del SNP rs4727338 (eje x) y el –log10 del valor de p (eje y) de su asociación con la DMO (eje y) está marcada con un cuadro de línea roja. Dicho SNP muestra la mayor significación en esta región. Los puntos de colores son las coordenadas del resto de SNPs estudiados en la región. Cada color indica un grado distinto de desequilibrio de ligamiento entre cada SNP y el SNP rs4727338. El gen FLJ42280 no está mostrado porque, en el momento en que se realizó el meta-análisis de GWAs, este gen todavía no estaba anotado en el genoma. Su localización entre SLC25A13 y SHFM1 se indica con un óvalo rojo. Esta figura es una modificación de la que se presenta en Estrada et al.[7]
En este contexto, el objetivo de este trabajo fue dar sentido a esta asociación mediante la determinación de cuál es la variante causal. ¿Es rs4727338 el SNP causal o hay otro SNP en desequilibrio de ligamiento con él que sea el verdadero SNP funcional? Para ello, hemos explorado la variabilidad genética de la región genómica donde se encuentra el gen FLJ42280 y hemos abordado la funcionalidad de estas variantes por enfoques diferentes. En primer lugar, por resecuenciación de la región en mujeres con DMO extremadamente alta o extremadamente baja para buscar variantes con una distribución desequilibrada entre los dos grupos; en segundo lugar, estudio bioinformático de la superposición de las variantes halladas con señales funcionales definidas en el proyecto ENCODE (The Encyclopedia of DNA Elements) y finalmente evaluación del posible papel como eQTLs de algunas de las variantes halladas.
Material y métodos
Selección de la muestra de estudio
La muestra de este estudio está formada por 100 mujeres de la cohorte BARCOS [10]. Esta cohorte está compuesta por unas 1.500 mujeres postmenopáusicas españolas monitorizadas en el Hospital del Mar de Barcelona. Las mujeres diagnosticadas de osteomalacia, de enfermedad de Paget, de algún trastorno metabólico o endocrino, o que estuvieran siguiendo una terapia de sustitución hormonal o tratadas con fármacos que pudieran afectar la masa ósea, fueron excluidas de la cohorte. Las mujeres con una menopausia temprana (antes de los 40 años) también fueron excluidas. La información recogida para cada muestra fue la DMO, la edad, la edad de menarquia, la edad de menopausia, los años desde la menopausia, el peso y la estatura. De cada paciente se obtuvieron muestras de sangre y consentimientos informados escritos, según las regulaciones del Comité Ético de Investigación Clínica del Parque de Salud Mar. La DMO (g/cm2) fue medida en el cuello del fémur y en la columna lumbar. Se utilizó un densitómetro de rayos X de energía dual para realizar las medidas.
Se seleccionaron dos grupos de 50 muestras con valores de DMO extremos, según el valor del Z-score. Concretamente, los grupos consistieron en las 50 muestras con el Z-score más alto (rango: de 2,98 a 0,73) y las 50 muestras con el Z-score más bajo (rango: de -2,41 a -4,26) de la cohorte BARCOS.
Preparación de las muestras genómicas
El ADN de cada mujer se extrajo a partir de muestras de sangre periférica. La concentración y la calidad de las muestras de ADN (ratios 260/280 y 260/230) se midieron por espectrofotometría en un aparato NanoDrop ND-1000 (NanoDrop Products). Para determinar la integridad del ADN, se analizaron 5 µl de cada muestra mediante electroforesis en gel de agarosa al 1%. Finalmente, las muestras se normalizaron a una concentración de 100 ng/µl.
Long-Range PCR (LR-PCR)
Se dividió una región genómica de 28 kb (que contiene el gen FLJ42280 [22 kb] junto con 3,8 kb de región flanqueante a 5’ y 2 kb de región flanqueante a 3’) en 7 fragmentos solapantes (Figura 2). Los tamaños y las coordenadas de estos 7 fragmentos y los pares de cebadores utilizados para amplificarlos, se muestran en la tabla 1. Los fragmentos, de entre 2 y 5 kb, se amplificaron mediante LR-PCR. Cada reacción de LR-PCR incluyó: 100 ng de ADN genómico, 5 µl de tampón Ex Taq “Magnesium +” (20 mM Mg++; Takara) x10, mezcla de dNTPs (a 2,5 mM cada uno), Ex Taq polimerasa (5 U/µl) y cebadores (20 µM), en un volumen final de 50 µl. Las reacciones se llevaron a cabo en un termociclador GeneAmp® PCR System 2700 (Applied Biosystems). Cada fragmento requirió unas condiciones de tiempo de elongación y temperatura de hibridación distintas. El número total de amplicones fue de 700 (100 muestras x 7 fragmentos). Se comprobó la cantidad y calidad de todos los amplicones mediante electroforesis en gel de agarosa al 1% p/v en tampón TBE x1.


Figura 2 En la región genómica de FLJ42280 estudios previos de GWA han identificado 4 SNPs que muestran asociación con la DMO, cuyos detalles se muestran en la tabla incluida en la figuraPara conocer mejor la variabilidad de este locus, la región se subdividió en 7 fragmentos solapantes (flechas amarillas) para su resecuenciación en mujeres con muy alta o muy baja DMO
Purificación y cuantificación de las muestras
Para eliminar los restos de los reactivos de la PCR, los productos de PCR se purificaron utilizando placas de filtro de 96 pocillos con un tamaño de poro adecuado (Pall Corporation). Se aplicó el vacío (Vacuum Manifold, Merck Millipore) y el ADN retenido en el filtro se resuspendió en 35 µl de agua milliQ. A continuación, los productos de PCR se cuantificaron mediante Quant-iT PicoGreen dsDNA Reagent and Kit (Life Technologies), según las instrucciones del fabricante. Brevemente, se construyó una curva estándar de concentraciones mediante medidas de emisión de fluorescencia a 520 nm después de haber excitado el ADN a 480 nm. La curva se utilizó posteriormente para calcular la concentración del ADN de las muestras.
Normalización de las concentraciones de las muestras y pooling
Las muestras en las placas se normalizaron a una concentración de 5 ng/µl y, a continuación, se mezcló en un solo tubo 5 µl de cada muestra de una placa (un tubo por placa) mediante el The epMotion® 5075 Liquid Handling Workstation (Eppendorf). Así, se obtuvieron 14 tubos con 250 µl cada uno, dos para cada fragmento de PCR (DMO alta y DMO baja). Los 14 pools se concentraron hasta 5 veces utilizando el Genevac EZ-2 evaporator (Genevac SP Scientific) y se cuantificó cada tubo mediante Qubit® 2.0 Fluorometer (Life Technologies). Finalmente, los fragmentos de PCR se mezclaron equimolarmente en dos tubos, uno para la DMO alta y otro para la DMO baja.
Secuenciación masiva en paralelo
La secuenciación masiva de las muestras se llevó a cabo en el Servicio de Genómica de los Centros Científicos y Tecnológicos de la Universidad de Barcelona, utilizando el GS 454 Junior System (Roche). Brevemente, se fragmentó el ADN por nebulización, se prepararon dos librerías marcadas con adaptadores (secuencias de 10 nucleótidos) distintos, una para cada grupo, que se mezclaron en un solo tubo. Seguidamente, se amplificó la mezcla por PCR de emulsión y la librería final se cargó en una placa picotiter dónde se llevó a cabo la pirosecuenciación. Se realizaron 4 carreras de secuenciación, correspondientes a 140 Mb de datos finales (35 Mb/carrera). Este volumen de datos proporciona una cobertura teórica de 40x para cada muestra inicial.
Procesamiento de los datos de secuenciación y selección de variantes
Las lecturas obtenidas de la secuenciación fueron preprocesadas en base a su calidad y se alinearon contra el genoma de referencia (GRCh37) utilizando el programa GS Mapper (Roche). Las lecturas se indexaron y filtraron utilizando SAMtools. Se detectaron las variantes presentes en los dos grupos mediante GATK utilizando parámetros de filtrado estándar [11]. Las variantes encontradas se priorizaron según los siguientes criterios: se seleccionaron las variantes con una cobertura de al menos 1.000 lecturas, presentes en un 1% de las lecturas y con un strand bias bajo. El número de lecturas de las variantes que pasaron los filtros fue normalizado por la cobertura y las variantes fueron clasificadas entre comunes (con una frecuencia mayor al 5%) y raras o de baja frecuencia (con una frecuencia menor al 5%).
Análisis funcional y estadístico de las variantes
Se compararon las frecuencias de cada variante entre los dos grupos mediante un test exacto de Fisher, aplicando la corrección de Bonferroni para comparaciones múltiples. El análisis funcional de las variantes consistió en mirar si estaban descritas en bases de datos como dbSNP y 1000 Genomas y, en caso afirmativo, buscar su MAF en la población europea e ibérica. Además, para las variantes exónicas se observó qué cambio de aminoácido suponían y su severidad predicha por SIFT, PolyPhen y Provean. Para las variantes intrónicas, se analizó la región que contiene la variante: lugares de hipersensibilidad a la DNAsa, unión de factores de transcripción, metilación del ADN, modificaciones de histonas, regiones reguladoras, etc. Todos estos datos fueron obtenidos de bases de datos y repositorios como Ensembl, UCSC Genome Browser, ENCODE, BioMart, MatInspector. También se utilizó HaploReg para buscar anotaciones de regulación. Finalmente, todas las variantes encontradas fueron analizadas con el Variant Effect Predictor de Ensembl y UCSC y con el SNP function prediction del Instituto Nacional de Ciencias de la Salud Ambiental (National Institute of Environmental Health Sciences) de los EE.UU.
Análisis del desequilibrio de ligamiento
Para calcular el desequilibrio de ligamiento entre todas las variantes de la región genómica de FLJ42280 se utilizaron los genotipos de los SNPs presentes en la región y los haplotipos de los individuos de HapMap fase 3. Para calcular tal desequilibrio y generar un gráfico se utilizó el software HaploView.
Análisis de eQTLs
Los SNPs que dieron significativos en los distintos GWAs y el SNP rs4613908 fueron evaluados como posibles eQTLs mediante dos aproximaciones: utilizando el portal del proyecto GTEx y utilizando los genotipos de esos SNPs en individuos de HapMap y los niveles de expresión de genes en cis en los mismos individuos. Concretamente, se obtuvieron los genotipos de los SNPs de 210 individuos no emparentados de la fase 1 y 2 de HapMap y los niveles de expresión de los genes SHFM1, SLC25A13 y DLX5 de una línea celular linfoblastoide de los mismos individuos obtenidos.
Resultados
Variantes halladas y pistas sobre su función
La región genómica de FLJ42280 (28 kb) se resecuenció masivamente en dos pools de DNA correspondientes a las 50 mujeres con mayor DMO y las 50 mujeres con menor DMO de la cohorte BARCOS (ver detalles en Material y Métodos), a una alta profundidad (alrededor 3.600x por grupo). Se comparó el número y la frecuencia de las variantes que se encontraron en cada grupo. Se identificó un total de 110 variantes, de las cuales 18 eran nuevas y 59 fueron variantes raras o de baja frecuencia (Tabla 2). Se observó que el número de variantes de baja frecuencia entre los dos grupos extremos era equilibrado. Así mismo, se observó que las diferencias de frecuencia de todas las variantes estaban por debajo de la potencia estadística del diseño, aunque 9 mostraron una tendencia.
Tabla 2 Número y localización de variantes de un solo nucleótido halladas en este estudio
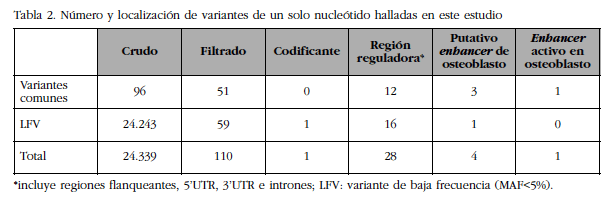
*incluye regiones flanqueantes, 5’UTR, 3’UTR e intrones; LFV: variante de baja frecuencia (MAF<5%).
Para cada variante, se analizó su superposición con elementos funcionales anotados en el genoma por el proyecto ENCODE. Cuatro de las variantes solaparon con posibles secuencias potenciadoras de la transcripción (o enhancers) de osteoblastos y de ellas una [SNP rs4613908; MAF(CEU)=0,39] solapó con un enhancer activo en osteoblastos (Figura 3).

Figura 3 A) Esquema de la región genómica de FLJ42280 y localización de algunas variantes prometedorasLas variantes comunes se señalan en rojo y las variantes de baja frecuencia (LFV) o raras en gris. Los cuadrados azules representan potenciadores (enhancers) de osteoblastos; el más oscuro representa un potenciador activo de los osteoblastos. B) Detalle del potenciador activo en osteoblastos. Se muestra el perfil de conservación de secuencia en vertebrados, los lugares de unión a factores de transcripción y las modificaciones de las histonas. Estos datos se extrajeron de UCSC Genome Browser -GRCh37- y de ENCODE (datos referidos a osteoblastos)
Análisis de desequilibrio de ligamiento
También se estudió el desequilibrio de ligamiento entre todas las variantes comunes en esta región. Se creó un gráfico de desequilibrio de ligamiento (LD) utilizando HaploView e información de haplotipos del proyecto HapMap (Figura 4) y se observó que hay un gran bloque de LD que incluye casi todo el gen (a excepción de la región 3’ UTR) y que por la parte upstream del gen se extiende 5 kb más allá de la región resecuenciada. También se constató que los SNPs rs4613908 y rs4727338 (meta-análisis de GWAs de Estrada et al. [7]) presentan una gran desequilibrio de ligamiento entre ellos.

Figura 4 Gráfico del desequilibrio de ligamiento presente en la región genómica de FLJ42280Se observa un bloque grande (24 kb) que incluye gran parte del gen excepto su región 3’UTR. Los 4 SNPs que se han asociado a DMO en los distintos GWAs están señalados con óvalos verdes. El SNP que solapa con el potenciador activo en osteoblastos está señalado con un óvalo morado. La línea horizontal azul indica la región genómica resecuenciada en este estudio.
Análisis de eQTLs
Para completar el análisis funcional, se realizó un análisis de eQTLs. Disponiendo de los genotipos de los cuatro SNPs asociados a la DMO, y del SNP rs4613908 de 210 individuos del proyecto HapMap y de los niveles de expresión génica de un array genómico en líneas linfoblastoides de estos mismos individuos, se determinó si los diferentes alelos o genotipos de los SNPs correlacionaban con los niveles de expresión génica de los genes situados en la región genómica de FLJ42280. Ninguno de los SNPs mostró influencia sobre los niveles de expresión de los genes SHFM1, SLC25A13 o DLX5 (en el array no hay información de niveles de expresión de FLJ42280). También se accedió a la base de datos GTEx para recabar información de eQTL para los mismos SNPs y el resultado fue negativo para todos ellos. Finalmente, se realizaron búsquedas de anotaciones de regulación en HaploReg. Este último análisis confirmó que la secuencia que rodea el SNP rs4613908 está altamente conservada entre los mamíferos y que en varios tipos celulares, incluyendo los osteoblastos primarios, contiene marcas de cromatina típicas de secuencias potenciadoras (H3K4me1, H3K27ac). Por otra parte, HaploReg destacó la alteración de motivos reguladores de este SNP y de rs10429035, pero no mostró ningún efecto de estos SNPs sobre la expresión génica.
Discusión
Se ha realizado un barrido exhaustivo de una región genómica (28 kb) en 7q21.3 que contiene varias señales muy fuertes de asociación entre 4 SNPs y la densidad mineral ósea [7,8,9]. Se ha querido conocer todas las variantes puntuales presentes en regiones codificantes (exones del gen FLJ42280) y no codificantes (intrones, 3’UTR, 5’UTR y flancos del gen) y evaluar el potencial funcional de estas variantes para pronosticar cuáles de ellas podrían ser las responsables de la asociación con la DMO. Se ha observado que la variante rs4613908 solapa con un potenciador génico (enhancer) activo en osteoblastos contenido en una secuencia con elevada conservación evolutiva. Dicho SNP (con sus dos variantes alélicas) podría estar afectando a la DMO por el hecho de alterar este potenciador génico. Queda por determinar cuál es el gen diana del mencionado potenciador.
Hasta la fecha, no nos constan otros trabajos de otros autores que hayan abordado la base funcional de la asociación con DMO de los SNPs situados en regiones no codificantes del gen FLJ42280. De hecho, este gen ha sido anotado recientemente en el genoma humano, de modo que cuando se detectó la asociación de los SNPs de la región, el gen todavía no constaba en el mapa de 7q21.3 y los SNPs quedaban entre los genes SLC25A13 y SHFM1 (Figura 1). Por ello, Estrada et al. [7] propusieron que la funcionalidad de la asociación podía estar relacionada con SLC25A13. Actualmente, FLJ42280 sigue siendo un gen anotado, con muy pocos datos experimentales que lo confirmen. Es pues muy probable que la función de los SNPs asociados a la DMO esté relacionada con otros genes. En este sentido, el gen SHFM1 se ha asociado a algunos casos hereditarios de malformación de mano hendida-pie hendido (Split hand and foot malformation 1; OMIM #183600) y el gen DLX5, situado a continuación, es de hecho el gen responsable de dicha enfermedad, dado que existen pacientes con mutaciones puntuales en DLX5 que cosegregan con la enfermedad [12]. Se han descrito una serie de potenciadores que afectan a la expresión de DLX5 en distintos tejidos y estadios del desarrollo y que se distribuyen a lo largo de varios cientos de kilobases. Estudios realizados en ratones y pez cebra han caracterizado estos potenciadores y han mostrado que funcionan durante el desarrollo [13,14]. Algunos de ellos muestran especificidad de tejido y correlacionan con determinados fenotipos presentes en pacientes con malformación de mano hendida-pie hendido portadores de varias anomalías cromosómicas (deleciones o translocaciones) que afectan a los potenciadores mencionados. Al colocar estos potenciadores de DLX5 sobre el mapa de la región 7q21.3, hemos visto con sorpresa que el SNP rs4613908, que acabamos de comentar como buen candidato funcional, se encuentra en uno de estos potenciadores (eDLX#18), situado a unas 500 kb de DLX5. El potenciador eDLX#18 se ha descrito como activo en los arcos branquiales en estadios embrionarios [13].
Existen evidencias de que DLX5 está involucrado en la determinación de la DMO [15], lo que nos hace proponer la hipótesis de que el potenciador eDLX#18 también es activo como un potenciador para DLX5 en osteoblastos de adultos y que nuestro SNP de interés es un eQTL en osteoblastos. Será muy necesario comprobar esta hipótesis mediante análisis de expresión de DLX5 en osteoblastos primarios y genotipación de rs4613908 de los mismos.

