










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.106 no.4 Madrid abr. 2014
La poliquistosis hepática del adulto (PHA) en España: análisis de una encuesta estructurada analizando la experiencia y actitud de los especialistas de digestivo españoles
Polycystic liver in the adult (PLA) in Spain: Analysis of a structured survey analysing the experience and attitude of gastroenterologists in Spain
Javier Ampuero1, Jesús M. Bañales2, Germán Soriano3, Javier Crespo4, José Luis Olcoz5, Moisés Diago6, José Luis Calleja7 y Manuel Romero-Gómez1
1Unidad de Gestión Médico-Quirúrgica de Enfermedades Digestivas y CIBERehd. Hospital Universitario de Valme. Universidad de Sevilla. Sevilla
2Instituto de Investigación Sanitaria Biodonostia. Hospital Universitario Donostia. Departamento de Enfermedades Hepáticas y Digestivas. IKERBASQUE (Fundación Vasca para la Ciencia). Universidad del País Vasco (UPV, EHU). CIBERehd. San Sebastián, Guipúzcoa
3Unidad de Hepatología y CIBERehd. Hospital de la Santa Creu i Sant Pau. Barcelona. Universitat Autònoma de Barcelona
4Servicio de Aparato Digestivo. Hospital Marqués de Valdecilla. Santander
5Servicio de Aparato Digestivo. Hospital de León. León
6Sección de Hepatología. Hospital General Universitario de Valencia. Valencia.
7Unidad de Hepatología y CIBERehd. Hospital Puerta de Hierro. Majadahonda, Madrid
Los autores agradecen a Pharma España el apoyo económico y logístico para la realización de la Encuesta Nacional y a la Junta Directiva de la SEPD por el aval científico para la puesta en marcha de esta iniciativa.
Dirección para correspondencia
RESUMEN
Antecedentes: la poliquistosis hepática del adulto (PHA) es una enfermedad rara caracterizada por el engrandecimiento crónico del hígado.
Objetivo: analizar la implicación, experiencia y actitud de los especialistas españoles en el diagnóstico, seguimiento y tratamiento de los pacientes con PHA.
Métodos: siete coordinadores del estudio contactaron cada uno con 15 especialistas de su entorno geográfico para participar en el estudio a través de una encuesta estructurada online.
Resultados: de los 105 clínicos contactados, 88 completaron el cuestionario, siguiendo una mediana de 3 pacientes por consulta, aunque 6 clínicos realizan seguimiento a > 20 pacientes con PHA. El seguimiento se realiza mayoritariamente en el departamento de hepatología (81 %) y/o gastroenterología (33 %). La ecografía hepática se usa para el diagnóstico (98 %) y seguimiento (97 %) de la mayoría de los pacientes. En el momento del diagnóstico, 76 % de los pacientes tenían < 50 años predominando las mujeres. El objetivo principal del manejo de los pacientes es el control sintomático. Un 28 % de los médicos prescriben tratamiento farmacológico, principalmente análogos de somatostatina, seguidos por inhibidores de mTOR. Un tercio de los clínicos indicó que tenían pacientes que habían recibido trasplante hepático y/o cirugía.
Conclusiones: la ecografía es la técnica de elección para el diagnóstico y seguimiento. Entre los clínicos que administran tratamiento farmacológico para el control sintomático, los análogos de somatostatina son los fármacos de elección. El uso poco frecuente de técnicas invasivas sugiere que los clínicos tienen una percepción bastante pobre de la utilidad de las distintas técnicas invasivas.
Palabras clave: Poliquistosis hepática. Enfermedad poliquística renal. Enfermedad poliquística hepática. Análogos somatostatina. Inhibidores mTOR. Trasplante hepático. Fenestración laparotómica o laparoscópica. Enfermedad poliquística hepática autosómica dominante (PCLD). Enfermedad poliquística renal autosómica dominante (PCKD).
ABSTRACT
Background: Polycystic liver in the adult (PLA) is a rare disease characterized by chronic liver enlargement.
Objective: To analyse gastroenterologists' involvement in, experience with, and attitude toward diagnosing, monitoring, and treating patients with PLA in Spain.
Methods: Each of seven study coordinators contacted 15 specialists in their geographic area about participating in the study via an online structured survey.
Results: Of the 105 clinics contacted, 88 completed the questionnaire, with a mean of 3 patients being followed per practice, although 6 clinics were following more than 20 patients with PLA. Patients were being followed mainly by the Department of Hepatology (81 %) and/or the Department of Gastroenterology (33 %). The majority of patients were diagnosed (98 %) and monitored (97 %) using liver ultrasound. When diagnosed, 76 % of patients were under 50 years of age, females predominating. The primary treatment objective for the patients was symptomatic management. Pharmacotherapy was prescribed by 28 % of physicians: Somatostatin analogues, primarily, followed by mTOR inhibitors. One-third of the clinics indicated that they had patients who had undergone liver transplant and/or surgery.
Conclusions: Ultrasound is the diagnosing and monitoring method of choice. Among the clinics using pharmacotherapy for symptomatic management, somatostatin analogues were the drugs of choice. These clinics' infrequent use of invasive procedures suggests that they perceive the various invasive techniques as not very effective.
Key words: Multiple hepatic cysts. Polycystic kidney disease. Polycystic liver disease. Somatostatin analogues. mTOR inhibitors. Liver transplant. Fenestration via laparotomy or laparoscopy. Autosomal-dominant polycistic liver-disease (PCLD). Autosomal-dominant polycistic kidney disease (PCKD)
Introducción
La poliquistosis hepática del adulto (PHA) es una enfermedad hereditaria y autosómica dominante que se caracteriza por la presencia de múltiples lesiones quísticas de origen biliar en más del 50 % del parénquima hepático, que pueden ser desde grandes masas, de 20 a 30 cm, a pequeños nódulos microscópicos (1,2). Es una enfermedad infrecuente con una incidencia estimada inferior al 0,01 % (3) y una prevalencia de 0,05-0,53 %, aunque la prevalencia de las mutaciones afecta a 1:600 (4). Con frecuencia los quistes hepáticos se presentan por primera vez en la cuarta década de vida, pero la historia natural de la enfermedad sugiere un crecimiento continuo ya que el número y el tamaño de los quistes van aumentando progresivamente con la edad (5). Por otro lado, existe una variante autosómica recesiva (ARPKD, enfermedad poliquística renal autosómica recesiva, por sus siglas en inglés, autosomal recessive polycystic kidney disease) caracterizada por dilataciones fusiformes no obstructivas de los conductos colectores renales y malformaciones de la vía biliar, con ectasia de los conductos biliares y fibrosis periportal.
La PHA es una enfermedad común a dos trastornos hereditarios autosómicos dominantes. Principalmente se asocia a enfermedad renal poliquística, lo que se conoce como enfermedad poliquística renal autosómica dominante (PCKD por sus siglas en inglés, polycystic kidney disease) (2,6). También puede presentarse de forma aislada sin asociarse a PCKD, enfermedad conocida como enfermedad poliquística hepática autosómica dominante (PCLD por sus siglas en inglés, polycystic liver disease). La PHA también puede estar asociada a poliquistosis en otros órganos, como el páncreas o el pulmón, pero en un porcentaje mucho menor.
Aunque hay varias publicaciones de casos clínicos de pacientes y familias con PHA en España (7-20), la epidemiología no está bien definida y el manejo de los pacientes con PHA en nuestro medio no está bien protocolizado. El objetivo de este estudio, avalado por Sociedad Española de Patología Digestiva, ha sido analizar la implicación y experiencia de los especialistas españoles en el diagnóstico, seguimiento y tratamiento de los pacientes con PHA con el fin de definir el manejo de estos pacientes en España y valorar si podría ser mejorado y/o consensuado.
Pacientes y métodos
La información sobre la actitud y la experiencia de los especialistas españoles en el manejo de la poliquistosis hepática en España se obtuvo a través de una encuesta estructurada que se completó online, con una duración aproximada de 15 minutos. El cuestionario de 49 preguntas estaba dividido en 6 secciones con varias preguntas en cada sección. La sección "Incidencia, prevalencia y derivación de pacientes" consistía en 12 preguntas sobre los pacientes con PHA tratados en la consulta del encuestado para estimar el número de pacientes y el manejo recibido por diferentes especialistas. La sección "Perfil de pacientes" consistía en 10 preguntas con la finalidad de detallar mejor al paciente español con PHA, preguntando por la caracterización de la PHA y su sintomatología asociada. La sección "Tratamiento quirúrgico poliquistosis hepática", con sus 4 preguntas abordaba la experiencia del encuestado con diferentes técnicas quirúrgicas en el tratamiento de la PHA. La sección "Tratamiento farmacológico poliquistosis hepática" tenía 7 preguntas para delinear la familiaridad del encuestado con los tratamientos farmacológicos disponibles, incluyendo nuevas terapias. Finalmente, las dos últimas secciones "Fuentes de información" y "Datos de clasificación" con 5 y 11 preguntas, respectivamente pretendían clasificar a los encuestados. La encuesta estuvo gestionada por 7 coordinadores y cada uno de los cuales contactó con 15 profesionales de su entorno geográfico. Una copia de la encuesta se encuentra disponible en el anexo a este artículo (Anexo).






Resultados
Encuesta
Casi la totalidad (104; 99 %) de los 105 clínicos contactados accedieron a participar y 88 (85 %) respondieron a la encuesta. La mitad de los encuestados se identificaron como hepatólogos (45; 51 %) y el resto como gastroenterólogos (34; 39 %), internistas, cirujanos y otras especialidades (9; 10 %). Aproximadamente la mitad de los encuestados llevaba más de 15 años tratando enfermedades hepáticas y el 31 % llevaba más de 20 años en esta profesión. Los participantes en el estudio representaban a la mayoría de Comunidades Autónomas, siendo la mayor participación de médicos de la provincia de Barcelona, seguidos por la provincia de Sevilla y Valencia (Fig. 1).

Demografía
De los 88 clínicos encuestados, 72 siguen a pacientes con PHA en su consulta. La mediana de seguimiento es de 3 pacientes por consulta, aunque en el caso de 6 de los 88 clínicos se realiza el seguimiento de más de 20 pacientes con PHA. De los especialistas encuestados, el 28 % recibe 1 nuevo paciente por año, el 16 % recibe 2 pacientes nuevos por año y el 15 % más de 2 pacientes nuevos por año. El porcentaje de pacientes con PCLD es del 44 % frente a un 56 % de pacientes con PCKD. En general, la PHA se suele diagnosticar antes de los 50 años, sin diferencias en la edad al diagnóstico entre los pacientes con PCLD y PCKD, y siendo dos tercios (65 %) de los sujetos mujeres. Pocos pacientes tienen un familiar directo con historia clínica de PHA. El fenotipo de múltiples quistes pequeños es el más frecuente. El 18 % de los hepatólogos respondieron que en los últimos 10 años menos de 5 pacientes fallecieron debido a la enfermedad o complicaciones relacionadas con la PHA. El 82 % restante no reflejó ninguna muerte relacionada a la PHA.
Flujo de pacientes
El paciente con PHA llega habitualmente diagnosticado al hepatólogo (73 % de los casos) derivado de otros departamentos clínicos; los pacientes son remitidos fundamentalmente desde nefrología (35 %), centros de atención primaria (33 %) o del departamento de gastroenterología (17 %). En el 75 % de los casos, el paciente con PHA diagnosticado por el hepatólogo no es derivado a ninguna otra especialidad para controlar la enfermedad. Asimismo, la mayoría de los pacientes diagnosticados de PCLD reciben seguimiento clínico por las unidades de hepatología (81 %) y/o gastroenterología (33 %). El seguimiento clínico de los pacientes con PCKD está peor definido, pero en el 28 % de los casos dicho seguimiento se realiza conjuntamente entre los servicios de nefrología y hepatología. También cabe remarcar que el 23 % de los pacientes con PCKD son vistos exclusivamente por el servicio de nefrología (Tabla I).

Síntomas
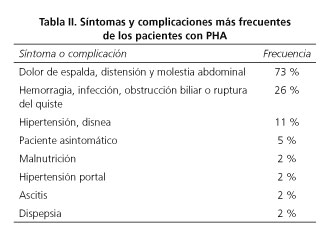
El dolor de espalda, la distensión y las molestias abdominales constituyen la sintomatología más frecuente (73 %), seguido por las complicaciones como hemorragias, infecciones, obstrucciones biliares o ruptura del quiste (26 % de los casos). En la experiencia de la mayoría de los clínicos (82,3 %), ninguno de sus pacientes había muerto por complicaciones asociadas a PHA (Tabla II).
Técnicas de diagnóstico y seguimiento
La técnica de elección para el diagnóstico y seguimiento de los pacientes con PHA es la ecografía hepática, utilizada en el 98 % de los casos para el diagnóstico y en el 97 % de los seguimientos. Otras técnicas incluyen la tomografía computarizada (TC), empleada en el 89 % de los diagnósticos y el 49 % de los seguimientos, y ya más distanciada, la resonancia magnética (RM), usada en el 58 % de los diagnósticos y el 30 % de los seguimientos.
Tratamiento
Globalmente, el objetivo principal del manejo de los pacientes con PHA es el control sintomático (78 %) seguido de la mejoría en la calidad de vida (12 %), la reducción (6,8 %) y estabilización (3,4 %) del volumen hepático.
Tratamiento farmacológico
Al igual que ocurre con el tratamiento quirúrgico, uno de los principales objetivos del tratamiento farmacológico es el control sintomático (Tabla III). En el último año, el 28 % de los clínicos empleó tratamiento farmacológico, siendo el tratamiento más frecuente los análogos de somatostatina seguido de los inhibidores de mTOR (del inglés, mammalian target of rapamycin - diana de rapamicina en células de mamífero). Aunque algunos clínicos no habían prescrito ningún fármaco, sí que reconocían el valor que pueden aportar el tratamiento con octeótrido, lanreótido, everolimus o sirolimus durante el manejo del paciente con PCLD. Del 18 % de los clínicos sin experiencia directa con el tratamiento farmacológico, el 40 % respondió que no consideraba oportuno prescribir tratamiento a sus pacientes con PHA debido al número importante de efectos adversos, seguido del 30 % que respondió que la razón por la cual no recomendaba tratamiento a sus pacientes era que estaban actualmente asintomáticos.

Tratamiento quirúrgico
En los últimos 10 años, un 40 % de los clínicos indicó que sus pacientes con PHA habían recibido un trasplante ortotópico de hígado (TOH). La enfermedad hepática terminal (end-stage liver disease en inglés) fue el motivo principal por el cual los pacientes recibían un TOH en el contexto de la PHA. El 14,8 % de los clínicos comentó que un paciente de su consulta había sido trasplantado, mientras que fueron dos para el 4,9 % de los clínicos y más de dos para el 16,4 %. Un tercio (33,8 %) de los clínicos indicó que tenía pacientes bajo su cuidado que habían sido intervenidos quirúrgicamente (13,1 % de los clínicos tenían un paciente, 14,8 % tenían dos pacientes y 4,9 % tenían más de dos pacientes que habían recibido cirugía), siendo la fenestración laparoscópica la técnica invasiva más frecuentemente indicada (75 %) y percibida como la de mejor ratio beneficio/riesgo (Tabla IV).

Discusión
En España, la PHA se percibe como una enfermedad rara, con la mayoría de médicos hepatólogos o gastroenterólogos tratando a menos de 5 pacientes con esta patología. Debido a esta cuestión, la encuesta presentada adquiere una gran relevancia ya que refleja la realidad y práctica clínica diaria de los profesionales en España. No obstante, hay que tener en cuenta que los resultados son aproximativos (no son derivados de un registro nacional de pacientes) y que existe una mayoría de encuestados de las provincias de Barcelona, Sevilla y Valencia.
En comparación con los hombres, las mujeres tienen mayor predisposición a desarrollar PHA masiva (> 15 quistes) y a tener quistes de mayor tamaño. Este carácter más dominante de la enfermedad en mujeres tiene un componente hormonal: el embarazo y el número de embarazos están correlacionados con un fenotipo más severo (6) y el uso de tratamiento con estrógenos en mujeres postmenopáusicas está asociado con un incremento del volumen hepático (21). La PHA es una enfermedad común a dos trastornos hereditarios autosómicos dominantes: la PCKD y la PCLD. La PCKD tiene su origen en mutaciones de dos genes: PKD1 (85 % de los casos) y PKD2 (15 % de los casos) (22), que codifican dos proteínas llamadas policistina 1 y 2, respectivamente. Estos genes se ubican en los cromosomas 16 y 4 (23). La manifestación extrarenal más común en PCKD es un hígado poliquístico. La prevalencia de PHA en pacientes con PCKD es del 58 % en pacientes de 15 a 24 años, 85 % en pacientes de 25 a 34 años y 94 % en pacientes de 35 a 46 años (24). Las mutaciones relacionadas con la PCLD se desconocen en un 80 % de los pacientes, teniendo su origen en el 20 % restante en la mutación de otros dos genes: PRKSCH y SEC63. El primer gen codifica para una proteína llamada hepatocistina, que se expresa en el retículo endoplásmico (25-27). En nuestro estudio hay un porcentaje similar de pacientes con PCLD y pacientes con PCKD. En España, el hepatólogo es el especialista que trata principalmente a los pacientes con PCLD. En cambio, aquellos que presentan PCKD son tratados en varios servicios, que en la mayoría de casos trabajan conjuntamente (especialmente, nefrología y hepatología). La mayoría de los pacientes llegan al hepatólogo ya diagnosticados y derivados por el nefrólogo o desde su médico de atención primaria. El diagnóstico se efectúa mediante pruebas de imágenes, como ecografía, TC (define mejor la extensión de la enfermedad hepática y la afección de los órganos adyacentes) y RM, aunque la ecografía es la técnica de elección para el diagnóstico y seguimiento.
La mayoría de pacientes están asintomáticos y preservan la función hepática (tanto la función sintética como la función excretora), por lo que el diagnóstico suele ser un hallazgo casual. Generalmente los quistes no producen síntomas excepto cuando alcanzan un gran tamaño o se complican por sangrado, infección, rotura, o malignidad (28). Cuando los quistes hepáticos son sintomáticos, la clínica se caracteriza por hepatomegalia dolorosa, distensión abdominal, sensación de saciedad o dolor lumbar (13). En nuestro estudio, los síntomas relacionados con la compresión que los quistes ejercen sobre el parénquima hepático adyacente y las estructuras vecinas fueron los predominantes (el dolor de espalda, la distensión y las molestias abdominales constituyeron el 73 %).
El principal objetivo del tratamiento farmacológico es el control sintomático, aunque el número total de pacientes que reciben tratamiento es bastante bajo. Aproximadamente sólo un tercio de los médicos encuestados en este estudio recomiendan tratamiento farmacológico para sus pacientes, siendo los análogos de somatostatina los fármacos de elección. Se ha demostrado que los quistes renales y hepáticos en pacientes con PCKD y con PCLD crecen en respuesta al cAMP (monofosfato cíclico de adenosina; cAMP por sus siglas en inglés, cyclic adenosine monophosphate) y que una disminución de los niveles de cAMP limita la progresión de la nefropatía o hepatopatía, sugiriendo que la inhibición de estos mecanismos podría ser de utilidad terapéutica. Los análogos de la somatostatina (como octreótido o lanreótido) reducen el cAMP intracelular y podrían impedir la acumulación de líquido en los quistes hepáticos (29,30). En un estudio reciente de 54 pacientes sintomáticos con PCLD y PCKD, el tratamiento con lanreótido durante 6 meses llevó a una disminución significativa del volumen hepático del 2,9 % en comparación con un incremento del 1,6 % en los pacientes que recibieron placebo (31). A pesar de esta evidencia, muchos de los médicos encuestados no prescriben estos fármacos por los efectos secundarios. Por otro lado, la interacción entre mTOR y la secreción de VEGF mediada por HIF-1 sugiere que el uso de inhibidores de mTOR podría ser eficaz en el tratamiento de PKCD (32). Además, el epitelio de los quistes hepáticos expresa altos niveles de mTOR sugiriendo que los inhibidores de mTOR pueden ser efectivos en el control del crecimiento de los quistes hepáticos (33). Estudios realizados en numerosos modelos experimentales de poliquistosis renal han mostrado que la rapamicina y everolimus (inhibidores de mTOR) retrasan el crecimiento de los quistes y protegen la función renal (33,34). Un meta-análisis reciente de la eficacia y seguridad del tratamiento con inhibidores mTOR en pacientes con PKCD concluyó que pueden disminuir el volumen renal sin una gran mejora en la función renal (35); sin embargo, no observó efecto sobre el volumen hepático en pacientes con PHA. En esta encuesta los inhibidores de mTOR eran el segundo tipo de fármaco reconocido y administrado por los encuestados, tras los análogos de la somatostatina.
En cuanto al tratamiento quirúrgico de los pacientes con PHA, la percepción de la utilidad de las distintas técnicas invasivas quirúrgicas es pobre, sobre todo por la transitoriedad de los beneficios. Aun así, un tercio de los clínicos encuestados han indicado que sus pacientes han recibido un trasplante hepático y/o tratamiento quirúrgico. El objetivo del tratamiento de la PHA es disminuir el tamaño de los quistes sin comprometer la función hepática, buscando que el paciente esté asintomático durante el periodo más largo posible (3). El tratamiento de estos pacientes consiste en la reducción del volumen de los quistes mediante el abordaje percutáneo (aspiración y alcoholización) o quirúrgico (fenestración por laparotomía o laparoscopia y/o resección hepática) (36,37). Por lo general, se intenta reservar la cirugía para los casos en que la terapia médica no es efectiva, aunque parece ser que en nuestro medio las técnicas quirúrgicas son el tratamiento de elección. La resección hepática parcial con fenestración de los quistes tiene cierto éxito en algunos pacientes con quistes sintomáticos masivos (30). A pesar de haberse realizado recientemente varias investigaciones, los resultados actuales no permiten decantarse claramente por la fenestración laparotómica o laparoscópica (3). No obstante, estos tratamientos son agresivos, caros y parcialmente efectivos ya que los síntomas suelen recurrir por el crecimiento de nuevos quistes o de los quistes ya tratados. Ocasionalmente, en los casos más graves es necesario realizar un trasplante hepático o trasplante combinado hepático y renal (36). La supervivencia, tanto del injerto como del paciente, es muy elevada en pacientes con PCLD que han recibido un trasplante hepático (38).
En conclusión, y reconociendo las posibles limitaciones de la encuesta, en nuestro medio la enfermedad PHA se trata de una patología poco frecuente, lo que hace que la experiencia clínica sea reducida. Además, su asociación con otros órganos implica que su manejo sea compartido entre diversos especialistas, destacando el papel del hepatólogo. El objetivo a conseguir con estos pacientes es controlar el tamaño de los quistes, siendo la ecografía la técnica de elección, y que permanezcan asintomáticos. Para ello, se emplean diversos tratamientos farmacológicos, como los análogos de la somatostatina o inhibidores de mTOR, quedando la opción quirúrgica para aquellos refractarios al tratamiento médico.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Manuel Romero-Gómez
UG Médico-Quirúrgica de Enfermedades Digestivas y CIBERehd
Hospital Universitario de Valme
Avda de Bellavista, s/n. 41014 Sevilla
e-mail: mromerogomez@us.es
Recibido: 02-09-2013
Aceptado 21-01-2014
Bibliografía
1. Vauthey JN, Maddern GJ, Kolbinger P, Baer HU, Blumgart LH. Clinical experience with adult polycystic liver disease. Br J Surg 1992;79:562-5. [ Links ]
2. Vauthey JN, Maddern GJ, Blumgart LH. Adult polycystic disease of the liver. Br J Surg 1991;78:524-7. [ Links ]
3. Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol 2007;13:5052-9. [ Links ]
4. Karhunen PJ, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clin Genet 1986;30:29-37. [ Links ]
5. Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology 2004;40:774-82. [ Links ]
6. Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology 1990;11: 1033-7. [ Links ]
7. Albandea Moreno C, Aguilar Urbano VM, Fernandez Perez F, Rivera Irigoin R, Gonzalo Marin J, Sanchez Cantos A. Polycystic liver disease. Rev Esp Enferm Dig 2009;101:495-7. [ Links ]
8. Campos Franco J, Otero Anton E, Gamborino Carames E, Martinez Castro J, Varo Perez E. Polycystic liver disease. Rev Esp Enferm Dig 2009;101:283-4. [ Links ]
9. Garcia-Gil FA, Guemes Sanchez A, Esteban Grau E, Lamata Hernandez F, Sousa Dominguez R, Serrano Aullo MT. Orthotopic liver transplantation for giant polycystic disease of the liver with terminal liver failure. Rev Esp Enferm Dig 2008;100:59-60. [ Links ]
10. Olivencia-Palomar P, Avila-Nasi S, Gonzalez-Soler R, Castro E, Lopez-Roses L. External and internal appearance of hepatorenal polycystic disease. Rev Esp Enferm Dig 2012;104:88-9. [ Links ]
11. Peces R, Cuesta-Lopez E, Peces C, Perez-Duenas V, Vega-Cabrera C, Selgas R. Octreotide reduces hepatic, renal and breast cystic volume in autosomal-dominant polycystic kidney disease. Int Urol Nephrol 2011;43:565-9. [ Links ]
12. Peces R, Drenth JP, Te Morsche RH, Gonzalez P, Peces C. Autosomal dominant polycystic liver disease in a family without polycystic kidney disease associated with a novel missense protein kinase C substrate 80K-H mutation. World J Gastroenterol 2005;11:7690-3. [ Links ]
13. Peces R, Gonzalez P, Venegas JL. Polycystic liver disease without autosomal dominant polycystic kidney disease. Nefrologia 2003;23:454-8. [ Links ]
14. Ramia JM, San Juan F, Orbis JF, Moya A, López-Andújar R, de Juan M, et al. Tratamiento de la poliquistosis hepática mediante trasplante hepático. Cir Esp 2004;76:358-62. [ Links ]
15. Romeo M, Tous F, Loza Y, Torres M. Tratamiento quirúrgico de la poliquistosis hepática sintomática, un problema infrecuente sin protocolizar. Med Clin (Barc) 2012;138:457-8. [ Links ]
16. Sanjuán F, Moya Á, López-Andújar R, Orbis F, De Juan M, Ramia JM, et al. Tratamiento de la poliquistosis hepática mediante transplante hepático Cir Esp 2004;76:358-62. [ Links ]
17. Selfa-Muñoz A, López-Segura RP, Eisman-Hidalgo M, Mundi Sánchez-Ramade JL. Poliquistosis hepática masiva. RAPD Online 2011;34:167-9. [ Links ]
18. Vall-Llovera J, Bosch A, Gil E, Pons L, Barba S, Palau M, et al. Poliquistosis hepática del adulto abscesificada. Cir Esp 2002;72:113-5. [ Links ]
19. Varona JF, Usandizaga I, Perez Maestu R, Marcos YRJ, Lozano F. 77 year old man with jaundice and pruritus. Rev Clin Esp 2006;206:197-8. [ Links ]
20. Tutor De Ureta P, Yebra Bango M. Renal polycystosis: A systemic disease?. Rev Clin Esp 2002;202:241-2. [ Links ]
21. Sherstha R, McKinley C, Russ P, Scherzinger A, Bronner T, Showalter R, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology 1997;26:1282-6. [ Links ]
22. Peters DJ, Sandkuijl LA. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol 1992;97:128-39. [ Links ]
23. Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 1999;353:103-7. [ Links ]
24. Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol 2006;1:64-9. [ Links ]
25. Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet 2003;33:345-7. [ Links ]
26. Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet 2004;36:575-7. [ Links ]
27. Li A, Davila S, Furu L, Qian Q, Tian X, Kamath PS, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet 2003;72:691-703. [ Links ]
28. Newman KD, Torres VE, Rakela J, Nagorney DM. Treatment of highly symptomatic polycystic liver disease. Preliminary experience with a combined hepatic. [ Links ]
29. Gevers TJ, Drenth JP. Somatostatin analogues for treatment of polycystic liver disease. Curr Opin Gastroenterol 2011;27:294-300. [ Links ]
30. van Keimpema L, de Man RA, Drenth JP. Somatostatin analogues reduce liver volume in polycystic liver disease. Gut 2008;57:1338-9. [ Links ]
31. van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, et al. Lanreotide reduces the volume of polycystic liver: A randomized, double-blind, placebo-controlled trial. Gastroenterology 2009;137:1661-8 e1-2. [ Links ]
32. Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, et al. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology 2010;138:360-71. [ Links ]
33. Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, et al. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008;19:631-8. [ Links ]
34. Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006;103:5466-71. [ Links ]
35. He Q, Lin C, Ji S, Chen J. Efficacy and safety of mTOR inhibitor therapy in patients with early-stage autosomal dominant polycystic kidney disease: A meta-analysis of randomized controlled trials. Am J Med Sci 2012;344:491-7. [ Links ]
36. Temmerman F, Missiaen L, Bammens B, Laleman W, Cassiman D, Verslype C, et al. Systematic review: The pathophysiology and management of polycystic liver disease. Aliment Pharmacol Ther 2011;34:702-13. [ Links ]
37. van Keimpema L, Drenth JP. Polycystic liver disease: A critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg 2011;253:419; author reply 20. [ Links ]
38. van Keimpema L, Nevens F, Adam R, Porte RJ, Fikatas P, Becker T, et al. Excellent survival after liver transplantation for isolated polycystic liver disease: An European Liver Transplant Registry study. Transpl Int 2011;24:1239-45. [ Links ]

