










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkFarmacia Hospitalaria
versión On-line ISSN 2171-8695versión impresa ISSN 1130-6343
Farm Hosp. vol.37 no.6 Toledo nov./dic. 2013
https://dx.doi.org/10.7399/FH.2013.37.6.1067
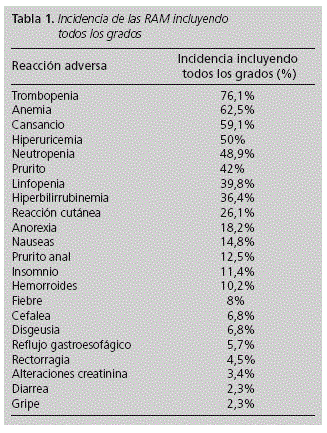
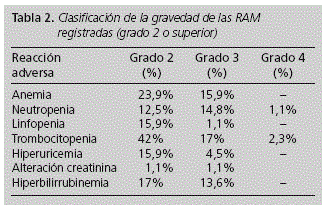
ORIGINALES Seguridad en la práctica clínica de la triple terapia con telaprevir en la hepatitis C crónica Safety in the clinical practica of the triple teraphy with telaprevir in chronic hepatitis C C. Sangrador Pelluz, F. J. Maiques Llácer y E. Soler Company Servicio de Farmacia. Hospital Arnau de Vilanova. Valencia. España. Dirección para correspondencia RESUMEN Objetivos: Estudiar la seguridad de la triple terapia con telaprevir y el momento de aparición de las RAM en el tratamiento de la hepatitis C. Palabras clave: Telaprevir; Seguridad; Triple terapia; Reacción adversa (RAM); Hepatitis C. ABSTRACT Purpose: To study the safety of triple therapy with telaprevir and the time of appearance of the RAM in the treatment of hepatitis C. Key words: Telaprevir; Safety; Triple therapy; Adverse drug reaction (ADR); Hepatitis C. Introducción La infección por el virus de la hepatitis C (VHC) es un problema de salud mundial, y actualmente es la primera causa de hepatopatía crónica y de trasplante hepático en los países occidentales1. Se estima que entre el 1 y el 5% de la población mundial está infectada por el VHC2,3. En Europa, los datos de prevalencia de infección por VHC oscilan 1,5 y el 3%4. La prevalencia de infección por VHC en portadores del VIH varía según distintos estudios entre un 45 y un 65%5. El principal factor que influencia la posibilidad de curación en pacientes con hepatitis C es el genotipo viral. La infección por genotipo 1 es la forma más común y supone el 75% de los infectados por VHC en España, mientras que la infección por genotipos 2 y 3 representa 20%-25% de los casos6. Hasta hace poco tiempo, el tratamiento convencional para la infección por VHC consistía en la combinación de interferón pegilado y ribavirina7,8 Sin embargo, a partir del 2011, fueron aprobados dos nuevos fármacos antivirales de acción directa, Telaprevir y Boceprevir, que actúan inhibiendo la proteasa de serina NS3*4 del VHC, enzima necesaria para la replicación del virus9, y que están indicados en combinación con peginterferon y ribavirina en el tratamiento de pacientes adultos con hepatitis C crónica genotipo 110. En Junio de 2011 se publicaron en New England Journal of Medicine los resultados de los ensayos clínicos de fase III ADVANCE11 y REALICE12 acerca de la terapia combinada con telaprevir. En ellos se describe una tasa de respuesta viral sostenida en torno al 75% en los pacientes naive y de aproximadamente el 50% en pacientes que habían fracasado a terapias previas. La incorporación del telaprevir al tratamiento de la hepatitis C crónica consigue incrementar la eficacia y acortar la duración del tratamiento, pero también aumentan notablemente los efectos adversos del mismo13. Las reacciones adversas (RAM) de grado 2 o superior notificadas con mayor frecuencia (incidencia > 5,0%) en los ensayos clínicos al asociar telaprevir fueron anemia, exantema, prurito, náuseas y diarrea10-12. En cuanto a las RAM clasificadas como graves (grado 3 o superior) más frecuentes (incidencia > 1,0%) fueron anemia,exantema, trombocitopenia, linfopenia, prurito y náuseas10-12. La incorporación de nuevos fármacos en la terapéutica actual, hace necesario la realización de estudios que evalúen su perfil de toxicidad en la práctica clínica. Conocer la seguridad de la terapia combinada con telaprevir, así como el momento de aparición de las RAM a lo largo del tratamiento, nos permitirá un mejor conocimiento y manejo del fármaco en el ámbito de la atención farmacéutica al paciente. Los objetivos del estudio son: -Estudiar la seguridad del tratamiento combinado con ribavirina, peginterferon alfa, y telaprevir para el tratamiento de la hepatitis C en la práctica clínica. -Conocer el momento de aparición de las RAM desde el inicio de tratamiento con telaprevir, para poder establecer futuras estrategias que nos ayuden a diseñar actuaciones en al ámbito de la seguridad al paciente. Material y métodos Estudio observacional retrospectivo de 1 año y 6 meses de duración (Enero 2012 - Junio 2013) realizado en nuestro Departamento de Salud. Población Los criterios de inclusión fueron: -Pacientes adultos infectados por VHC genotipo 1, tanto monoinfectados como coinfectados por VIH, que hubieran finalizado las 12 semanas de tratamiento con telaprevir en combinación con peginterferon y ribavirina durante el periodo de estudio. Método A partir de los programas informáticos empleados en el Servicio de Farmacia para la atención a pacientes externos, Farmasyst® y Orion Clinic®, se recogieron las variables necesarias para caracterizar a los pacientes, y aquellas referentes al tratamiento recibido. Se recogieron las siguientes variables relacionadas con los pacientes: edad, sexo, genotipo del VHC, grado de fibrosis, tipo de paciente (naive o pretratados), y coinfección con VIH. Referente al tratamiento, se registraron: reacciones adversas recogidas en la historia clínica, necesidad de transfusión, de factores estimuladores de eritropoyesis, o de factores estimuladores de colonias de granulocitos, reducciones de dosis de peginterferon y/o ribavirina, y suspensiones de tratamiento. Evaluación de la seguridad La seguridad se valoró identificando el tipo y la gravedad de los eventos adversos desencadenados por el tratamiento. La clasificación de las RAM se realizó siguiendo los criterios de la División del SIDA (DAIDS, versión 1.0)14. La clasificación de la toxicidad cutánea se realizó de acuerdo a la ficha técnica del telaprevir, clasificándose en leves, moderadas o graves10. Evaluación del tiempo hasta la aparición de las RAM a telaprevir Mediante la revisión de la historia clínica informatizada se registraron las RAM analíticas detectadas tras el inicio de la triple terapia con telaprevir. Se midió: -Tiempo hasta la aparición de la RAM: tiempo que transcurre desde el inicio de la triple terapia con telaprevir hasta la aparición de la RAM independientemente del grado -Tiempo hasta la aparición de la RAM de mayor grado: tiempo transcurrido desde el inicio del tratamiento hasta que se evidencie el mayor grado de dicha RAM -Tiempo hasta la necesidad de tratamiento de soporte: tiempo desde el inicio del tratamiento hasta la prescripción de eritropoyetina exógena o factores estimuladores de colonias. Los datos se analizaron mediante el programa estadístico SPSS® versión 15.0. Se realizó un análisis estadístico descriptivo, incluyendo medidas de tendencia central y dispersión, y frecuencias absolutas y relativas. Para el tiempo hasta la aparición de la toxicidad se empleó un análisis de supervivencia de Kaplan Meier donde el tiempo se midió como días, y se expresó como mediana e intervalo. Resultados Se incluyeron 88 pacientes (78% hombres) con una edad media de 51,6 ± 8,6 años. Respecto al tipo de paciente: 40,9% eran pacientes naive, 35,2% recaedores, 12,5% respondedores parciales, y 11,4% respondedores nulos. El 40,9% de los pacientes estaba coinfectado con el VIH. El 62,5% eran pacientes con infección por VHC genotipo 1B, y el 37,5% presentaban genotipo 1A. Respecto al grado de fibrosis, el 75% de los pacientes presentaban un grado de fibrosis F4 (cirrosis grado A según la clasificación Child-Pugh), el 18,2% grado F3, y el 6,8% grado F2. La incidencia de las RAM observadas incluyendo todos los grados se recoge en la tabla 1. La clasificación de la gravedad de las RAM (grado 2 o superior) de acuerdo a la clasificación DAIDS se recoge en la tabla 2. En cuanto a la toxicidad cutánea, el 12,5% de los pacientes presentaron reacciones dérmicas leves, el 10,2% moderadas y el 3,4% de tipo grave. Respecto al tratamiento, el 51,1% de los pacientes requirió ajuste de dosis de peginterferon y/o ribavirina, siendo el 38,6% de los casos reducciones de ribavirina y en el 12,5% reducciones de ribavirina y peginterferon. Durante la triple terapia, el 13,6% de los pacientes requirieron transfusiones de sangre, el 28,4% eritropoyetina exógena y el 1,1% estimuladores de colonias de granulocitos. El 8% de los pacientes requirió ingreso hospitalario motivado por la toxicidad del tratamiento. El tratamiento fue suspendido debido a su toxicidad en el 6,8% de los pacientes: 3 por toxicodermia grave, 2 por toxicidad hematológica grave, y 1 por emesis grave. Los tiempos hasta la aparición de las alteraciones analíticas y de su progresión hasta la máxima toxicidad se recogen en la tabla 3. El tiempo hasta la necesidad de tratamiento de soporte fue de 58 días (22-82) para la eritropoyetina exógena, y de 22 días en el paciente que requirió factores estimuladores de colonias. Discusión La triple terapia con telaprevir se ha convertido en la actualidad en uno de los tratamientos estándar para los pacientes con hepatitis por VHC genotipo 1. Dicho tratamiento ha sido evaluado en dos ensayos clínicos pivotales11,12 demostrando mayores tasas de curación que la terapia de referencia (peginterferon y ribavirina), aunque con mayor toxicidad. Sin embargo, en la actualidad no existen datos publicados a cerca de la toxicidad de la nueva terapia combinada con telaprevir en la práctica clínica habitual, así como de su momento de aparición durante el tratamiento. La distribución de nuestra población en cuanto al sexo y edad media fue similar a la publicada en los ensayos clínicos11,12, sin embargo, en cuanto al genotipo del VHC, se observó una prevalencia superior del genotipo 1b (62,5%) respecto a los estudios publicados (41-45%)11,12. Además, la tasa de cirrosis hepática en nuestra población (75%) fue superior a la referida en los ensayos clínicos (21-26%)11,12, hecho que podría deberse a que en nuestra población se incluyeron pacientes coinfectados con VIH, que requerían un grado de fibrosis igual o superior a F3 para iniciar el tratamiento con telaprevir de acuerdo a las normas de nuestra Comunidad Autónoma4. En un análisis conjunto de cinco estudios de fase II y III 11-18 que incluyó 1346 pacientes que recibieron tratamiento combinado con telaprevir, se observó una incidencia de exantema del 55% y de prurito del 51%, siendo en nuestro caso del 26,1% y del 42% respectivamente. Al igual que se describe en los principales estudios10, aproximadamente el 90% de las RAM dermatológicas registradas en nuestro trabajo fueron de tipo leve-moderado, sin embargo, se observó una incidencia de toxicodermia grave inferior a la publicada (3,4% vs 4,8%)11,12. La incidencia de anemia en nuestro estudio (incluyendo todos los grados) fue del 62,5% frente al 32,1% referida en los ensayos clínicos fase II-III 11,12,16-18. Además, en dichos estudios se observaron valores de hemoglobina <10 g/dl en el 34% de los pacientes que recibieron telaprevir10, dato inferior al obtenido en nuestro caso (39,8%). Respecto a las RAM graves (G3 o superior) registradas en nuestro estudio, la incidencia de linfopenia fue similar a la observada en los principales ensayos clínicos (1,1% vs 0,6%)11,12. En cuanto a los síntomas anorrectales, la incidencia observada en nuestro estudio fue del 12,5% para el prurito anal y del 10,2% en el caso de las hemorroides, frente al 6% y 12% señalada en los ensayos clínicos11,12,16-18. La suspensión del tratamiento con telaprevir por RAM graves fue del 6,8% en nuestro trabajo, frente al 10% referenciado en los trabajos publicados11,12. En cuanto a la tasa de transfusión de concentrados de hematíes, fue considerablemente superior en nuestro estudio (13,6%) respecto a la publicada en los ensayos clínicos (2,5%)10. Además, en dichos estudios no se permitieron medicamentos estimulantes de la eritropoyesis y solo se emplearon en el 1% de los pacientes11,12. El ajuste de dosis de ribavirina durante el tratamiento con telaprevir fue superior en nuestro estudio respecto a los datos publicados (51,1% vs 21,6%)11,12. Nuestro estudio señala la aparición de la trombocitopenia y la hiperbilirrubinemia en su mayor grado de toxicidad de manera temprana. Sin embargo, en el resto de RAM se observaron periodos de progresión más prolongados hasta alcanzar el máximo grado de toxicidad. El descenso de los niveles de hemoglobina tuvo lugar en las primeras cuatro semanas de tratamiento, alcanzando sus valores más bajos al final del periodo de tratamiento con telaprevir, hecho que coincide con los datos publicados10. En cuanto a la terapia de soporte, el tiempo hasta la necesidad de tratamiento con eritropoyetina exógena se correspondió bien con el momento de aparición de anemia de mayor grado. Respecto a las limitaciones de nuestro estudio hay que señalar que se trata de un estudio retrospectivo con datos obtenidos de las historias clínicas, encontrando en ocasiones una falta de información por incompleta cumplimentación de las mismas. En cuanto a la comparación de nuestros resultados frente a los señalados en otras publicaciones, es conveniente tener en cuenta que nuestra población difiere principalmente de las de los ensayos clínicos en que no es homogenea, ya que incluye a diferente tipo de pacientes (naive y pretratados) y coinfectados con VIH. En conclusión, los resultados de nuestro estudio señalan un perfil de toxicidad superior al descrito en los principales ensayos clínicos. En concreto, se observa una mayor incidencia de toxicidad hematológica, principalmente en cuanto a la anemia y trombocitopenia. El conocimiento del momento de aparición de la toxicidad al tratamiento nos permite predecir un rango de tiempo de mayor probabilidad de aparición de estas RAM, de manera que se puedan diseñar estrategias multidisciplinares para poder prevenir y mejorar el manejo de las mismas. Bibliografía 1.Liang T, Rehermann B, Seeff L, Hoofnagle J. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med. 2000;132:296-305. [ Links ] 2.Cornberg M, Razavi H, Alberti A, et al. A systematic review of hepatitis C virus epidemiology in Europe, Canada and Israel. Liver Int. 2011;2:30-60. [ Links ] 3.European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatitis C virus infection Journal of Hepatology 2011;55:245-64. [ Links ] 4.Resolución del Secretario Autonómico de la Agencia Valenciana de Salud de declaración como MAISE de Telaprevir y Boceprevir y establecimiento de directrices y criterios clínicos de utilización en pacientes con hepatitis crónica C. Versión 3 (actualización 10/06/ 2013) (Consulta 01/07/2013). Disponible en: http://www.san.gva.es/web/dgfps/programa-paise [ Links ] 5.Castilla, J. y De la Fuente, L.Trends in the number of human immunodeficiency virus infected persons and AIDS cases in Spain: 1980-1998]. Med. Clin. 2000;115:85-9. [ Links ] 6.Romero Gómez M, Lacalle Remigio JR. Tratamiento de la hepatitis crónica C por genotipos 2 y 3: revisión sistemática. Gastroenterología y hepatología. 2006;2:139-45. [ Links ] 7.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958-65. [ Links ] 8.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL Jr, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975-82. [ Links ] 9.Díaz Ferrer, J. Actualización en el tratamiento de la Hepatitis C. Acta Méd. Peruana. 2012;4:208-12. [ Links ] 10.Ficha técnica de Incivo (Consulta 01/07/2013). Disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002313/WC500115529.pdf [ Links ] 11.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, et al. Telaprevir for Previously Untreated Hepatitis C Virus Infection. N Engl J Med. 2011;364:2405-16. [ Links ] 12.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R. Telaprevir for Retreatment of HCV Infection. N Engl J Med. 2011;364:2417-28 [ Links ] 13.Boceprevir y telaprevir en el tratamiento de la hepatitis C. Boletín Farmacoterapéuitco Andaluz no2 (Vol.28).Marzo-Abril 2012. (consulta 4/7/2013) Disponible en: http://www.cadime.es/es/boletin_terapeutico_andaluz.cfm?bid=122 [ Links ] 14.Criterios de la División del SIDA (DAIDS, versión 1.0). (consulta 5/7/2013) Disponible en: http://www.hptn.org/webdocuments/hptn046/ssp/appendices/appendixe-toxicitytablesdaidsaegradingtablefinaldec2004.pdf [ Links ] 15.Comité Asesor de fármacos antivíricos de la FDA. Documento de instrucciones sobre telaprevir. Abril 2011. (consulta 4/7/2013). Disponible en: http://www.fda.gov/downloads/AdvisoryCommittees/ComitteesMeetingMaterials/Drugs/AntiviralDrugsAdvisoryCommittee/UCM252562.pdf [ Links ] 16.McHutchison JG, Everson GT, Gordon SC, Jacobson IM, Sulkowski M, Kauffman R, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009; 360:1827-38. [ Links ] 17.McHutchison JG, Manns MP, Muir AJ, Terrault NA, Jacobson IM, Afdhal NH, et al. Telaprevir for previously treated chronic HCV infection. N Engl J Med. 2010;362:1292-303. [ Links ] 18.Hezode C, Forestier N, Dusheiko G, Ferenci P, Pol S, Goeser T, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 2009;360:1839-50. [ Links ] Recibido: 24/07/2013.
Método: Estudio observacional retrospectivo (Enero 2012-Junio 2013) de los pacientes con VHC genotipo 1 que hubieran finalizado las 12 semanas de triple terapia con telaprevir. Se recogieron las variables necesarias para caracterizar a los pacientes, y aquellas referentes al tratamiento recibido. La clasificación de las RAM se realizó según criterios de la División del SIDA versión 1.0.
Resultados: Se incluyeron 88 pacientes (78% hombres), 75% pacientes cirróticos. El 40,9% estaba coinfectado con VIH. Las principales RAM (incidencia > 40%) incluyendo todos los grados: toxicidad hematológica, cansancio, hiperuricemia, hiperbilirrubinemia y prurito. Las RAM graves (incidencia > 15%): trombocitopenia, anemia y neutropenia. El 3,4% presentó toxicodermia grave. El 51,1% requirió ajuste de dosis de ribavirina, 13,6% transfusiones de sangre, y 28,4% eritropoyetina exógena. El 8% requirió ingreso hospitalario motivado por la toxicidad del tratamiento. El tratamiento fue suspendido por toxicidad en el 6,8% de los pacientes: 3 por toxicodermia grave, 2 por toxicidad hematológica grave, y 1 por emesis grave. La trombocitopenia y la hiperbilirrubinemia se registraron de manera temprana en su mayor grado de toxicidad, mientras que el resto de RAM presentaron periodos de progresión más prolongados.
Conclusiones: El estudio señala un perfil de toxicidad superior al descrito en los ensayos clínicos, principalmente en cuanto a toxicidad hematológica, y permite predecir un rango de tiempo de mayor probabilidad de aparición de las RAM.
Method: A retrospective observational study (January 2012-June 2013) of patients with HCV genotype 1 who had completed 12 weeks of triple therapy with telaprevir. The following variables were needed to characterize patients, and those relating to the treatment received. The classification of ADR was performed according to criteria of the SIDA Division 1.0.
Results: We included 88 patients (78% male), 75% cirrhotic patients. 40.9% were coinfected with HIV. The main ADR (incidence > 40%) including all grades: haematological toxicity, fatigue, hyperuricemia, hyperbilirubinemia and pruritus. Serious ADR (incidence > 15%): thrombocytopenia, anemia and neutropenia. 3.4% had severe toxicodermia. 51.1% required ribavirin dose adjustment, blood transfusions 13.6%, and 28.4% exogenous erythropoietin. The 8% required hospitalization motivated by treatment toxicity. The treatment was stopped for toxicity in 6.8% of patients: 3 severe toxicodermia, 2 severe haematological toxicity, and 1 severe emesis. Thrombocytopenia and hyperbilirubinemia occurred at an early stage in its higher degree of toxicity, while the other ADR presented progression longer periods.
Conclusions: The study shows a superior toxicity profile to that described in clinical trials, mainly with regard to hematologic toxicity, and predicts a time range of highest probability of occurrence of ADR.

![]() Dirección para correspondencia:
Dirección para correspondencia:
Cristina Sangrador Pelluz
E-mail: sangrador_cri@gva.es
Aceptado: 07/10/2013.

